1.本发明关于用于制备苯并前列环素类似物的新颖方法和中间物,以及由其制得的苯并前列环素类似物。
背景技术:
2.由于前列环素的发现,已研发出多种化学稳定和代谢稳定的苯并前列环素类似物作为临床上有效的抗血栓剂。其中,由toray industries inc.研发的贝前列素(beraprost)为最引人注目的化合物的一种。市售贝前列素钠(beraprost sodium)为外消旋化合物,亦即,以下四种异构物的混合物:
3.贝前列素钠
[0004][0005]
镜像异构性纯的贝前列素-314d为贝前列素的药理活性异构物,目前正在进行临床试验,用于治疗肺高血压和血管疾病。
[0006]
贝前列素或贝前列素-314d的合成已揭示于先前技术中,诸如us5202447、cn106478572、wo2017/174439、us8779170、us9334255、us9765047、us8779170、tetrahedron lett.1990,31,4493.,和org.lett.2017,19,1112,但其效率相当低。
[0007]
us7345181揭示藉由共轭加成反应以更高效率合成贝前列素的途径,如以下流程a中所示,其自环戊烯酮a1开始经由共轭加成以形成中间物a2(步骤1):
[0008]
流程a
[0009][0010]
如流程a中所示,具有9-酮基的中间物a2与三(仲丁基)硼氢化锂(l-selectride)反应以获得具有9α-羟基的中间物a3(步骤2)。然后,中间物a3经历sn2反应以将9α-羟基转化为9β-氯基以便形成中间物a4(步骤3)。接着,移除中间物a4的苯酚的甲氧基甲基保护基和羟基的叔丁基二甲基硅烷基保护基(步骤4),且进行分子内环化反应以获得中间物a6(步骤5)。
[0011]
然而,流程a中所描绘的方法需要至少五个步骤以自环戊烯酮a1形成苯并前列环素的三环结构。因此,需要发展和研发一种用于在工业中形成苯并前列环素的三环结构的高效率的共轭加成方法。
技术实现要素:
[0012]
本发明的主要目的为提供用于产生具有较高非镜像异构和镜像异构纯度的苯并前列环素类似物和其中间物的更高效共轭加成方法,由此形成具有高纯度的产品。
[0013]
本发明的一方面提供一种式1的外消旋或光学富集的环戊烯酮:
[0014][0015]
其中p为h或p1;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1为羟基保护基。
[0016]
本发明的另一方面提供一种制备富含(r)-对映异构体且光学纯度为至少95%对映异构体过量(e.e.)的式(r)-1”的化合物的方法,
[0017][0018]
其中x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基。
[0019]
本发明的另一方面提供一种用于制备式4的苯并前列环素类似物的方法,
[0020][0021]
其中p为h、p1或p2;r2为h或c
1-4
烷基;r3为c
1-7
烷基、c
2-7
炔基、芳基或芳氧基,其各自未经取代或经c
1-4
烷基、卤素或三卤甲基取代;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1或p2为羟基保护基。
[0022]
本发明的另一方面提供一种用于制备式4a的光学富集苯并前列环素类似物的方法,
[0023][0024]
其中p为h、p1或p2;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1和p2为羟基保护基。
[0025]
本发明的另一方面提供一种用于制备式4b的外消旋苯并前列环素类似物的方法,
[0026][0027]
其中p为h、p1或p2;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1和p2为羟基保护基。
[0028]
本发明的另一方面提供一种式4a”的光学富集中间物,其中y为cl、br或i,其用于制备贝前列素-314d,
[0029][0030]
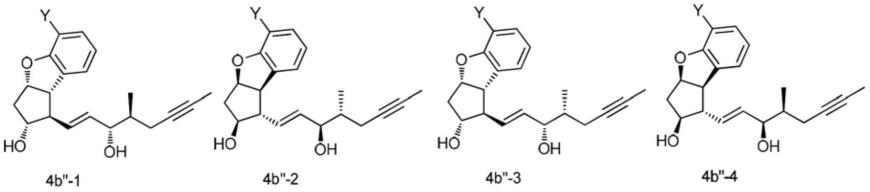
本发明的再一方面提供一种式11,15-同侧4b”的外消旋中间物,其中y为cl、br或i,其用于制备贝前列素,且至少包含式4b
”‑
1、式4b
”‑
2、式4b
”‑
3和式4b
”‑
4的四种异构物:
[0031]
11,15-同侧4b"
[0032]
具体实施方式
[0033]
定义
[0034]
当与术语「包含」结合用于申请专利范围和/或说明书中时,使用词语「一(a/an)」可意谓「一个(one)」,但其亦与「一或多个」、「至少一个」和「一个或超过一个」的含义相符。尽管本发明支持仅指替代方案和「和/或」的定义,但除非明确指示为仅指替代方案或除非替代方案相互排斥,否则在申请专利范围中使用术语「或」用于意谓「和/或」。在本技术案通篇中,术语「约」用于指示值,其包括装置、用以测定所述值的方法或研究个体当中存在的变化导致的固有误差变化。
[0035]
如本说明书和申请专利范围中所使用,术语「包含(comprising)」(和包含的任何形式,诸如「包含(comprise/comprises)」)、「具有(having)」(和具有的任何形式,诸如「具有(have/has)」)、「包括(including)」(和包括的任何形式,诸如「包括(includes/include)」)或「含有(containing)」(和含有的任何形式,诸如「含有(contains/contain)」)为包括性或开放的,且并不排除其他未列出的要素或方法步骤。
[0036]
除非另外规定,否则本文所用的术语「烷基」是指含有1至30个碳原子、优选1至20个碳原子且最佳1至12个碳原子的直链或分支链烃基,诸如甲基、乙基、异丙基、叔丁基和其类似基团;或具有3至20个碳原子且优选3至10个碳原子的环状饱和烃基,诸如环丙基、环戊基、环己基、基和其类似基团。
[0037]
除非另外规定,否则本文所用的术语「炔基」是指含有2至30个碳原子,且优选3至20个碳原子和一或多个碳-碳参键的直链或分支链烃基,诸如戊炔基、丙炔基和其类似基
团;或具有6至20个碳原子和一或多个碳-碳参键的环状不饱和烃基。
[0038]
除非另外规定,否则本文所用的术语「芳基」是指具有6至30个碳原子,且优选6至20个碳原子的单环或多环芳族烃基,诸如苯基、萘基、蒽基、菲基和其类似基团。
[0039]
除非另外规定,否则本文中所用的术语「芳烷基」是指含有1至20个碳原子和一或多个如上文所描述的芳基的直链或分支链烃,诸如苯甲基、二苯甲基、茀基甲基和其类似者。
[0040]
上文所提和的烷基、炔基、芳基和芳烷基中的每一者可视情况经一或多个选自由以下组成的群的取代基取代:卤素、烷基、芳基、烷氧基、芳氧基、硫烷氧基、硫芳氧基、烷胺基、芳胺基、氰基、烷氧羰基、芳基羰基、芳基胺基羰基、烷胺基羰基和羰基,或选自由以下组成的群的杂环基:吡啶基、噻吩基、呋喃基、咪唑基、吗啉基、恶唑啉基、吡啶基、哌嗪基、四氢吡喃基、吡咯烷基、吡咯烷酮基和类似基团。
[0041]
除非另外说明,否则术语「羟基保护基」具有习知地定义于有机合成化学中的含义,亦即能够保护化合物的羟基基团或部分以抵抗化学反应的攻击的基团。羟基保护基的实例包括但不限于甲氧基甲基、甲氧基硫代甲基、2-甲氧基乙氧基甲基、双(2-氯乙氧基)甲基、四氢吡喃基、四氢硫代吡喃基、4-甲氧基四氢吡喃基、4-甲氧基四氢硫代吡喃基、四氢呋喃基、四氢硫代呋喃基、1-乙氧基乙基、1-甲基-1-甲氧基乙基、三苯甲基、烯丙基、苯甲基、经取代的苯甲基和sirarbrc,其中ra、rb和rc各自独立地为未经取代或经取代的烷基或未经取代或经取代的芳基,诸如c1-4烷基、苯基、苯甲基、经取代苯基和经取代苯甲基。
[0042]
在本说明书通篇给出的化合物的描述中,非楔形黑体键()或楔形黑体键()意谓在纸平面上方突起的键。非楔形切割键()或楔形切割键()意谓在纸平面下方突起的键。楔形黑体键()和楔形切割键()用于绝对组态;非楔形黑体键()和非楔形切割键()表示相对组态和外消旋特征;和波浪键()意谓几乎一半在纸平面上方突起和一半在纸平面下方突起的键。
[0043]
式1'的外消旋环戊烯酮的合成途径
[0044]
本发明提供一种式1'的外消旋环戊烯酮的制备方法:
[0045][0046]
其中p1为羟基保护基,x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基。
[0047]
根据本发明,式1'化合物可根据流程b中所示的反应制备。如流程b中所示,式5化合物(其中x为cl、br或i)与二异丙胺基锂(lda)反应以经由区域选择性锂化使其转化成有机锂中间物,然后以二甲基甲酰胺(dmf)甲酰化以获得式6化合物,其中x如上文所定义(步骤1)。随后,经由1,2-加成反应用2-锂呋喃处理式6化合物以得到式7化合物(步骤2)。此后,式7的化合物经皮安卡泰利重排(piancatelli rearrangement)(步骤3)和异构化(步骤4),得到式1”的环戊烯酮。最后,保护式1”化合物的二级羟基以获得式1'化合物,其中p1为羟基
保护基,且x如上文所定义(步骤5)。在一些实施态样中,可对式6、7、8、1”或1'的化合物进行铃木(suzuki)交叉偶合反应,其中x为cl、br或i,以形成式6、7、8、1”或1'的化合物,其中x为-ch2ch2ch2coor1,且其中r1为c
1-7
烷基或c
7-11
芳烷基,
[0048]
流程b
[0049][0050]
本发明亦提供一种式1的外消旋或光学富集化合物:
[0051][0052]
其中p为h或p1;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1为羟基保护基。
[0053]
式1”的外消旋环戊烯酮的对掌性解析
[0054]
根据本发明,根据流程c中所示的反应,式1”的外消旋环戊烯酮可容易地对掌性解析以形成富含(r)-对映异构体且具有高光学纯度的环戊烯酮。如流程c中所示,式1”的外消旋化合物(其中x为cl、br或i)藉由使用第一脂肪酶经由对映体选择性酯化来解析,形成式(s)-1”的未反应醇与式8的酯的混合物(步骤1)。随后,式(s)-1”的未反应醇可容易地移除,例如藉由经由管柱纯化使其自混合物分离,且随后继续进行光延反应(mitsunobu reaction),以得到相应式8化合物。或者,混合物可直接经受光延反应(步骤2)以将式(s)-1”的未反应醇转化为式8化合物。最后,藉由使用化学水解反应或酶裂解反应,使式8化合物去酰化,以形成富含(r)-对映异构体且具有高光学纯度的式(r)-1”化合物,
[0055]
流程c
[0056][0057]
在流程c的步骤1中,式1”的环戊烯酮的对映体选择性酯化用式d的酰基供体(其中r4和r5独立地为h或c
1-6
烷基)在第一脂肪酶存在下进行,其中酰基供体优先与(r)形式的环戊烯酮反应,由此产生基本上由式8的光活性酯和式(s)-1”的未反应醇组成的混合物,
[0058][0059]
在一些实施态样中,适合的第一脂肪酶为市售的,且可衍生自无色杆菌属(achromobacter sp.)、柱状念珠菌(candida cylindracea)、产碱杆菌属(alcaligenese sp.)、南极念珠菌(candida antarcitica)、洋葱假单胞菌(pseudomonas cepacia)、洋葱伯克霍尔德氏菌(burkholderia cepacian)、施氏假单胞菌(pseudomonas stutzri)或其混合物,优选产碱杆菌属、柱状念珠菌、南极念珠菌、洋葱伯克霍尔德氏菌或施氏假单胞菌,且最佳洋葱伯克霍尔德氏菌。适合的酰基供体包括但不限于乙酸乙烯酯、乙酸异丙烯酯、戊酸乙烯酯、戊酸异丙烯酯、丁酸乙烯酯、丁酸异丙烯酯和其混合物,且乙酸乙烯酯尤其优选。此外,对映体选择性酯化反应可在单一有机溶剂或有机溶剂的混合物中进行,所述有机溶剂诸如己烷、环己烷、甲苯、四氢呋喃、甲基乙基酮、甲基异丁基酮、醚、异丙基醚、甲基异丙基醚、叔丁基甲基醚和其混合物。适当的反应温度在约5℃至约50℃范围内,优选在环境温度下。
[0060]
流程c的步骤2是关于光延反应。由于醇和酯的极性不同,因此式(s)-1”的未反应醇可藉由管柱纯化容易地自流程c的步骤1获得的混合物分离出来,进行光延反应,从而形成式8的酯。在一些实施态样中,不必将式(s)-1”和式8化合物与混合物分离,且混合物可直接进行光延反应以将式(s)-1”的未反应醇转化成式8的酯。在此步骤中,混合物中几乎100%的式(s)-1”的醇可转化为式8的酯。
[0061]
在光延反应中,可用式r4cooh的酰氧基供体(其中r4为h或c
1-6
烷基)处理式(s)-1”的未反应醇,以在适合溶剂中在偶氮二羧酸二烷基酯和三烷基膦/三芳基膦存在下将其转化成式8化合物。适合的偶氮二羧酸二烷基酯包括但不限于偶氮二甲酸二甲酯(dmad)、偶氮二甲酸二乙酯(dead)、偶氮二甲酸二异丙酯(diad)、偶氮二甲酸二叔丁酯(dtbad)、偶氮二甲酸二苯甲基酯(dbad)、偶氮二甲酸双三氯乙基酯(btcead)、偶氮二甲酸二对氯苯甲基酯
(dcad)、偶氮二甲酸二4-硝基苯甲基酯(dnad)、偶氮二甲酸二环戊基酯(dcpad),和其混合物;且优选偶氮二甲酸二乙酯、偶氮二甲酸二异丙酯、偶氮二甲酸二苯甲基酯和其混合物。适合的三烷基膦/三芳基膦包括但不限于三正丁基膦、三苯膦和其混合物;且优选为三苯膦。光延反应中的适合溶剂包括但不限于四氢呋喃、甲苯、苯、二甲基甲酰胺、乙醚、乙腈、二氯甲烷和其混合物。光延反应优选在介于约-30℃至约70℃范围内的合适温度下,优选在环境温度下进行。
[0062]
流程c的步骤3是关于去酰化反应,诸如化学水解反应或酶裂解反应。在一些实施态样中,去酰化反应为在醇系统中使用酸催化剂的化学水解反应。适合的酸催化剂包括但不限于磷酸、对甲苯磺酸、氢溴酸、盐酸、硝酸、硫酸和其混合物。醇系统中的适合醇包括但不限于甲醇、乙醇、丙醇、异丙醇、丁醇、异丁醇和其混合物。举例而言,在硫酸和甲醇存在下进行式8化合物的化学水解反应。
[0063]
在一些实施态样中,流程c的步骤3中的去酰化反应为酶裂解反应。酶裂解反应可在适当温度下在第二脂肪酶存在下于适合的有机溶剂或水性系统中进行,以获得式(r)-1”的化合物。适合的第二脂肪酶为市售的,且可衍生自无色杆菌属、产碱杆菌属、南极念珠菌、洋葱假单胞菌、施氏假单胞菌、假单胞菌属或其混合物;且优选无色杆菌属、产碱杆菌属、南极念珠菌、假单胞菌属或其混合物;且最佳南极念珠菌。
[0064]
根据本发明,出于所得式(r)-1”化合物的光学纯度的目的,监测去酰化反应。在一些实施态样中,去酰化藉由hplc使用对掌性管柱监测,且优选在所得化合物的光学纯度降低至约95%e.e.,优选约99%e.e.,且更优选约99.9%e.e.时藉由移除第二脂肪酶停止。在一些实施态样中,可诸如藉由管柱层析在去酰化反应后移除式8的未反应酯和其对映异构体。根据本发明,所产生的式(r)-1”的化合物的光活性为至少约95%e.e.,优选为至少约99%e.e.,且最佳为至少约99.9%e.e.。
[0065]
因此,本发明提供一种制备富含(r)-对映异构体且光学纯度为至少95%e.e.的式(r)-1”的化合物的方法,
[0066][0067]
其中x为cl、br或i,所述方法包含以下步骤:
[0068]
(1)对式1”化合物进行对映体选择性(r)-酯化:
[0069][0070]
所述酯化用式d的酰基供体:
[0071][0072]
其中r4和r5独立地为h或c
1-6
烷基,和第一脂肪酶进行,以形成(s)-醇与(r)-酯的混合物;
[0073]
(2)视情况移除所述(s)-醇;和
[0074]
(3)使所述(r)-酯去酰化。
[0075]
在一些实施态样中,可对式(r)-1”的化合物进行铃木交叉偶合反应,其中x为cl、br或i,以形成式(r)-1”的化合物,其中x为-ch2ch2ch2coor1,且其中r1为c
1-7
烷基或c
7-11
芳烷基。
[0076]
制备式4的苯并前列环素类似物
[0077]
本发明亦提供一种式4的苯并前列环素类似物的制备方法:
[0078][0079]
其中p为h、p1或p2;r2为h或c
1-4
烷基;r3为c
1-7
烷基、c
2-7
炔基、芳基或芳氧基,其各自未经取代或经c
1-4
烷基、卤素或三卤甲基取代;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1和p2为羟基保护基。
[0080]
根据本发明,式4化合物可根据流程d中所示的反应制备。如流程d中所示,式4化合物的合成开始于式1'化合物,其中步骤1是关于1,4-加成反应,步骤2是关于还原反应,和步骤3是关于分子内环化反应,
[0081]
流程d
[0082][0083]
随后描述的细节可用于制备本发明的苯并前列环素类似物的方法中的任一者。在流程d的步骤1中,式2的环戊烯酮(其中p、x、r2和r3如上文所定义)是藉由式1'的环戊烯酮
(其中p1和x如上文所定义)与衍生自式l1、式l2或式l3的化合物(其中y为cl、br或i;p2、r2和r3如上文所定义)的铜酸盐的ω侧链单元的偶合反应来制备。在一些实施态样中,反应在约-100℃至约-20℃范围内的温度下进行,优选在约-80℃至约-40℃下进行,
[0084][0085]
流程d的步骤2是关于酮还原反应。在步骤2中,用还原剂将式2化合物的c9-羰基还原为α-羟基。适合的还原剂包括但不限于氢化双(2-甲氧乙氧基)铝钠、氢化二异丁基铝、三-叔丁氧基氢化铝锂、三烷基硼氢化锂、三烷基硼氢化钾、三烷基硼氢化钠和其混合物;优选三-二级丁基硼氢化锂(l-selectride)、三-二级丁基硼氢化钠(n-selectride)、三-二级丁基硼氢化钾(k-selectride)、三戊基硼氢化锂、三戊基硼氢化钾和其混合物;且在此步骤中三-二级丁基硼氢化锂(l-selectride)作为还原剂为更优选的。
[0086]
如流程d的步骤3中所示,藉由式3化合物在适合的碱条件存在下的分子内环化反应制备式4的苯并前列环素类似物。在一些实施态样中,分子内环化反应藉由在介于约0℃至约120℃范围内的温度下在适合溶剂中使用适合碱来达成。适合碱包括但不限于氢化钠、氢化钾、氢化锂、叔丁醇钾、丁基锂和其混合物。适合溶剂包括但不限于四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚、二甲基甲酰胺、n,n'-二甲基丙烯脲、1,2-二甲氧基丙烷、甲苯和其混合物。
[0087]
与流程a中所展示的需要五(5)个步骤以形成苯并前列环素的三环结构的习知共轭加成反应相比,本发明的方法仅需要三(3)个步骤以形成苯并前列环素的三环结构。本发明的方法为更高效的共轭加成方法。
[0088]
在一些实施态样中,可对所获得的式2、3或4化合物进行去保护反应以移除羟基保护基。
[0089]
在一些实施态样中,可对式2、3或4的化合物进行铃木交叉偶合反应,其中x为cl、br或i,以形成式2、3或4的化合物,其中x为-ch2ch2ch2coor1,且其中r1为c
1-7
烷基或c
7-11
芳烷基。铃木交叉偶合反应优选在式br
2-ch2ch2ch2coor1的烷基硼烷(其中r为烷基)存在下进行,其由3-丁烯酸烷基(r1)酯(其中r1为c
1-7
烷基或c
7-11
芳烷基)与硼烷试剂制备,所述硼烷试剂诸如9-硼双环[3.3.1]壬烷(9-bbn)、二-二级异戊基硼烷(disiamylborane)、二异戊基硼烷、儿茶酚硼烷、二异松蒎甲硼烷(diisopinocamphenylborane)、二环己基硼烷、双(频哪醇基)二硼烷和其混合物。铃木交叉偶合反应亦可在钯催化剂、配位体和碱存在下,在介于约50℃至约60℃范围内的温度下,在氮气或氩气下进行。适合钯催化剂包括但不限于pd(pph3)4、pd(dppf)2cl
2-dcm、pd(dppf)2cl2、pd(oac)2、pd2(dba)2、双(eta3-烯丙基-mu-氯钯(ii)和其混合物。在一些实施态样中,钯催化剂可用配位体处理以形成用于促进交叉偶合反应的反应性的钯错合物。适合的配位体包括但不限于pph3、asph3、p(ome)3、(n-bu)3p、dppe、dppp、二环己基-(2,6-二甲氧基联二苯-2基)膦和其混合物。适合碱可提高烷基硼烷对形成pd卤化物错合物的反应性以促进交叉偶合速率,所述适合碱包括但不限于li2co3、na2co3、k2co3、cs2co3、naome、k3po4、t-buon、t-buok、k3po4、naoh和其混合物。在一些实施态样中,铃木交叉偶合反应在pd(dppf)2cl
2-dcm、asph3和k3po4存在下在60℃下于四氢呋喃溶
剂中进行。
[0090]
因此,本发明提供一种用于制备式4化合物的方法:
[0091][0092]
其中p为h、p1或p2;r2为h或c
1-4
烷基;r3为c
1-7
烷基、c
2-7
炔基、芳基或芳氧基,其各自未经取代或经c
1-4
烷基、卤素或三卤甲基取代;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1和p2为羟基保护基,所述方法包含以下步骤:
[0093]
(1)使式1'化合物:
[0094][0095]
其中p1和x如上文所定义,与衍生自式l1、式l2或式l3化合物的铜酸盐反应:
[0096][0097]
其中y为cl、br或i;p2、r2和r3如上文所定义,以形成式2化合物:
[0098][0099]
其中p、x、r2和r3如上文所定义;
[0100]
(2)还原所述式2化合物的酮以形成式3化合物:
[0101][0102]
其中p、x、r2和r3如上文所定义;
[0103]
(3)进行所述式3化合物的分子内环化反应,以形成式4化合物:
[0104][0105]
其中p、x、r2和r3如上文所定义;
[0106]
(4)视情况进行去保护反应以移除所述羟基保护基;和
[0107]
(5)视情况进行式2、3或4的化合物的铃木交叉偶合反应,其中x为cl、br或i,以形成式2、3或4的化合物,其中x为-ch2ch2ch2coor1且r1如上文所定义。
[0108]
式4a的光学富集苯并前列环素类似物的合成途径
[0109]
本发明进一步提供一种式4a的光学富集化合物的制备方法:
[0110][0111]
其中p为h、p1或p2;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1和p2为羟基保护基。
[0112]
如流程e中所描绘,式4a化合物的合成类似于流程d中所展示的式4化合物的合成。式4a化合物的合成开始于式(r)-1'的光学富集化合物,其中p1和x如上文所定义;和开始于式l
1a
、式l
2a
或式l
3a
的光学富集化合物,
[0113][0114]
其中y为cl、br或i;且p2为羟基保护基,流程e
[0115]
[0116]
在流程e的步骤1中,藉由式(r)-1'的光学富集化合物与衍生自式l
1a
、l
2a
或式l
3a
的光学富集化合物的铜酸盐的ω侧链单元的偶合反应来制备式2a的化合物。随后,使式2a化合物经受还原反应(步骤2)和分子内环化反应(步骤3)以获得式4a化合物,其中x和p如上文所定义。
[0117]
在一些实施态样中,可对所获得的式2a、3a或4a化合物进行去保护反应以移除羟基保护基。在一些实施态样中,可经由铃木交叉偶合反应使式2a、3a或4a的化合物(其中x为cl、br或i)转化为式2a、3a或4a的化合物,其中x为-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基。
[0118]
因此,本发明提供一种用于制备式4a的光学富集化合物的方法:
[0119][0120]
其中p为h、p1或p2;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1和p2为羟基保护基;
[0121]
所述方法包含以下步骤:
[0122]
(1)使式(r)-1'的光学富集化合物:
[0123][0124]
其中p1和x如上文所定义,与衍生自式l
1a
、式l
2a
或式l
3a
的光学富集化合物的铜酸盐反应:
[0125][0126]
其中y为cl、br或i;且p2为羟基保护基,以形成式2a化合物:
[0127][0128]
其中p和x如上文所定义;
[0129]
(2)还原所述式2a化合物的酮以形成式3a化合物:
[0130][0131]
其中p和x如上文所定义;
[0132]
(3)进行所述式3a化合物的分子内环化反应,以形成式4a化合物:
[0133][0134]
其中p和x如上文所定义;
[0135]
(4)视情况进行去保护反应以移除所述羟基保护基;和
[0136]
(5)视情况进行所述式2a、3a或4a的化合物的铃木交叉偶合反应,其中x为cl、br或i,以形成所述式2a、3a或4a的化合物,其中x为-ch2ch2ch2coor1且r1如上文所定义。
[0137]
式4b的苯并前列环素类似物的合成途径
[0138]
本发明进一步提供一种式4b的化合物的制备方法:
[0139][0140]
其中p为h、p1或p2;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1和p2为羟基保护基。
[0141]
式4b化合物可根据流程f中所示的反应制备:
[0142]
流程f
对映异构体为错配对,因此其偶合反应不倾向于产生11,15-反侧异构物。根据本发明,非所需11,15-反侧异构物(亦称为15-差向异构物)可藉由管柱层析或结晶分离。
[0150]
在一些实施态样中,藉由去除式2b、3b或4b化合物的保护基,其中p为羟基保护基,可获得式2b、3b或4b化合物,其中p为h。
[0151]
在一些实施态样中,可经由铃木交叉偶合反应使式2b、3b或4b的化合物(其中x为cl、br或i)转化为式2b、3b或4b的化合物,其中x为-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基。
[0152]
因此,本发明提供一种用于制备式4b外消旋化合物的方法:
[0153][0154]
其中p为h、p1或p2;x为cl、br、i或-ch2ch2ch2coor1,其中r1为c
1-7
烷基或c
7-11
芳烷基;且p1和p2为羟基保护基,所述方法包含以下步骤:
[0155]
(1)使式1'外消旋化合物:
[0156][0157]
其中p1和x如上文所定义,与衍生自包含式l
1b
、式l
2b
或式l
3b
的化合物的外消旋和非镜像异构混合物的铜酸盐反应:
[0158][0159]
其中y为cl、br或i;p2如上文所定义,以形成式2b化合物:
[0160][0161]
其中p和x如上文所定义;
[0162]
(2)还原所述式2b化合物的酮以形成式3b化合物:
[0163][0164]
其中p和x如上文所定义;
[0165]
(3)进行所述式3b化合物的分子内环化反应,以形成式4b化合物:
[0166][0167]
其中p和x如上文所定义;
[0168]
(4)视情况进行去保护反应以移除所述羟基保护基;和
[0169]
(5)视情况进行所述式2b、3b或4b的化合物的铃木交叉偶合反应,其中x为cl、br或i,以形成式2b、3b或4b的化合物,其中x为-ch2ch2ch2coor1且r1如上文所定义。
[0170]
本发明进一步提供式11,15-同侧4b”的外消旋中间物,其中y为cl、br或i,其用于制备贝前列素
[0171]
11,15-同侧4b"。
[0172]
根据本发明,式11,15-同侧4b”的外消旋中间物至少包含式4b
”‑
1、式4b
”‑
2、式4b
”‑
3和式4b
”‑
4的四个11,15-同侧异构物:
[0173][0174]
其中y为cl、br或i。
[0175]
式11,15-同侧4b”的化合物具有极佳结晶度,且因此可易于藉由结晶纯化以移除非所需的11,15-反侧异构物。因此,非所需11,15-反侧异构物的量可在所得中间混合物中降低至不超过约5%、1%或0.1%。
[0176]
本发明亦提供式4a”的新颖的光学富集中间物,其中y为cl、br或i,其用于制备贝前列素-314d,
[0177][0178]
根据本发明,式4a”化合物具有极佳结晶度,且因此可容易藉由自极性/非极性溶剂混合物中进行的结晶方法结晶。适合的极性/非极性溶剂系统包括但不限于乙酸乙酯/正己烷、乙酸乙酯/正庚烷、mtbe/正庚烷和iproac/正庚烷混合物。此外,由前述反应产生的杂质可藉由重复结晶方法降低至不超过约5%、1%或0.1%,以得到高纯度化合物。
[0179]
本文中所揭示和主张的所有化合物和/或方法可根据本发明在无不当实验的情况下制成和执行。虽然已根据优选实施例描述本发明的化合物和方法,但熟习此项技术者应清楚变化可在不背离本发明的概念、精神和范畴的情况下应用于本文所描述的组合物和/或方法中和步骤中或方法的步骤顺序中。对熟习此项技术者显而易见的所有此类类似取代和修改均视为在由随附权利要求书所定义的本发明的精神、范畴和概念内。
[0180]
实例
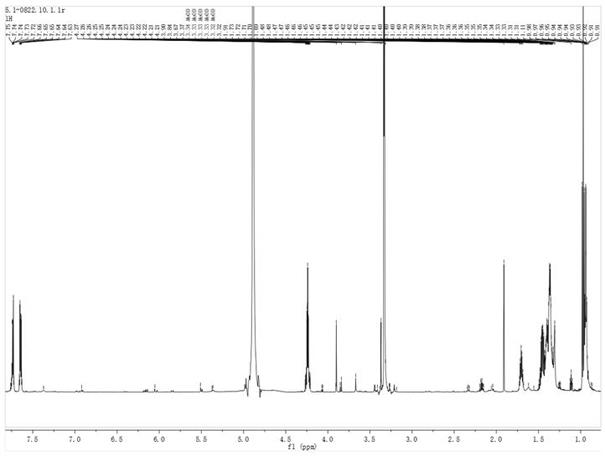
[0181]
实例1
[0182]
(3-溴-2-氟苯基)(呋喃-2-基)甲醇
[0183][0184]
在氮气下在-50℃下,向二异丙胺(43.3g,0.43莫耳)于无水thf(220ml)中的溶液中逐滴添加含1.6m(270ml,0.43莫耳)n-buli的thf。使反应混合物升温至-10℃。在一小时后,使反应混合物冷却至-70℃。藉由滴液漏斗向其中缓慢添加1-溴-2-氟苯(50.0g,0.29莫耳)于无水thf(200ml)中的溶液,且保持反应温度在-70℃与-65℃的间。随后,在-70℃下搅拌反应混合物30分钟。随后,藉由滴液漏斗向其中缓慢添加糠醛(28.8g,0.30莫耳)于无水thf(29ml)中的溶液,且保持反应温度在-70℃与-65℃的间。藉由tlc监测确认反应完成。用饱和氯化铵水溶液(400ml)淬灭反应混合物。对反应混合物进行相分离,且用乙酸乙酯(200ml)萃取水层。将有机层合并且经无水na2so4干燥。滤出固体且在真空下蒸发掉有机溶剂,得到78g粗标题化合物。
[0185]
实例2
[0186]
5-(3-溴-2-氟苯基)-4-羟基环戊-2-烯酮
[0187][0188]
向(2-溴-3-氟苯基)(呋喃-2-基)甲醇(78g,0.29莫耳,来自实例1)于thf/h2o(780ml,thf/h2o=8/1)的混合物中的溶液中,添加p-tsoh-h2o(54.95g,0.29莫耳),且在60℃下搅拌反应混合物。在反应完成后,藉由10%nahco3水溶液(800ml)淬灭混合物,且对反
应混合物进行相分离,且用乙酸乙酯(500ml)萃取水层。将有机层合并且经无水na2so4干燥。滤出固体且在真空下蒸发掉有机溶剂,得到82.5g粗标题化合物。
[0189]
实例3
[0190]
2-(3-溴-2-氟苯基)-4-羟基环戊-2-烯酮
[0191][0192]
在室温下,向粗5-(3-溴-2-氟苯基)-4-羟基环戊-2-烯酮(82.5g,来自实例2)于甲苯(825ml)中的溶液中,添加三乙胺(30.9g,0.305莫耳)和水合氯醛(5.05g,30.5mmol)。藉由tlc监测确认反应完成。混合物用10%nacl
(水溶液)
(800ml)洗涤且经无水na2so4干燥。滤出固体且在真空下蒸发掉有机溶剂。藉由硅胶层析使用己烷和乙酸乙酯的混合物作为梯度溶离剂来纯化粗产物。标题化合物的产量为28.81g(45%,3个步骤)。
[0193]1h-nmr(400mhz,cdcl3):δ7.813-7.824(m,1h),7.716-7.756(m,1h),7.512-7.552(m,1h),7.036-7.079(m,1h),5.099-5.114(m,1h),2.931-2.993(m,1h),2.455-2.507(dd,1h)。
[0194]
实例4
[0195]
2-(3-溴-2-氟苯基)-4-((四氢-2h-吡喃-2-基)氧基)环戊-2-烯酮
[0196][0197]
用二氯甲烷(510ml)溶解2-(3-溴-2-氟苯基)-4-羟基环戊-2-烯酮(51.0g,143.6mmol,来自实例3)。随后,向其中添加乙酸(740mg,9.4mmol)和二氢吡啶(24g,285.3mmol)。在室温下搅拌反应混合物。藉由tlc监测确认反应完成。用10%nahco
3(水溶液)
(510ml)淬灭反应混合物且对反应混合物进行相分离。收集有机层且将其经无水na2so4干燥。滤出固体且在真空下蒸发掉有机溶剂,得到粗化合物。藉由硅胶层析使用己烷和乙酸乙酯的混合物作为梯度溶离剂来纯化粗产物。标题化合物的产量为52g(79%)。
[0198]1h-nmr(400mhz,cdcl3):δ7.891-7.944(d,1h),7.746-7.791(m,1h),7.507-7.544(t,1h),7.036-7.075(t,1h),5.019-5.050(m,1h),4.817-4.884(m,1h),3.870-3.960(m,1h),3.549-3.604(m,1h),2.875-2.985(m,1h),2.489-2.667(m,1h),1.539-1.862(m,6h)。
[0199]
实例5
[0200]
乙酸(r)-3-(3-溴-2-氟苯基)-4-侧氧基环戊-2-烯-1-基酯
[0201][0202]
用甲苯(250ml)稀释2-(3-溴-2-氟苯基)-4-羟基环戊-2-烯酮(25g,92.6mmol,来自实例3),且随后向其中添加脂肪酶-sl(2.5g)和乙酸乙烯酯(25g)。在室温下搅拌反应混
合物2小时。藉由对掌性hplc管柱监测反应完成。自反应混合物过滤脂肪酶-sl,且浓缩滤液,得到粗混合物(31.25g)。
[0203]
随后,将三苯膦(15.18g,57.8mmol)和乙酸(3.47g,57.8mmol)添加至含所述粗混合物的甲苯(300ml)中。在室温下搅拌反应混合物,直至将三苯膦溶解于反应混合物中为止。随后向其中缓慢添加偶氮二甲酸二异丙酯。藉由tlc监测确认反应完成。在真空下进一步移除反应溶剂且用乙酸乙酯(150ml)和正己烷(450ml)的混合物悬浮残余物。滤出固体且在真空下蒸发滤液。藉由硅胶层析使用己烷和乙酸乙酯的混合物作为梯度溶离剂来纯化粗产物。标题化合物的产量为26.6g(92%)。
[0204]1h-nmr(400mhz,cdcl3):δ7.809-7.819(m,1h),7.736-7.769(m,1h),7.497-7.537(m,1h),7.020-7.060(m,1h),5.883-5.910(m,1h),2.964-3.028(m,1h),2.477-2.528(m,1h),2.090(s,3h)。
[0205]
实例6
[0206]
(r)-2-(3-溴-2-氟苯基)-4-羟基环戊-2-烯酮
[0207][0208]
将脂肪酶-435(3.0g)添加至乙酸(r)-3-(3-溴-2-氟苯基)-4-侧氧基环戊-2-烯-1-基酯(15g,48mmol,来自实例5)于甲基异丁基酮(mibk,150ml)中的溶液中。在室温下搅拌反应混合物四小时。自反应混合物过滤脂肪酶-435,且浓缩滤液,得到粗化合物。藉由硅胶层析使用己烷和乙酸乙酯的混合物作为梯度溶离剂来纯化粗产物。标题化合物的产量为10.65g(83%)。
[0209]1h-nmr(400mhz,cdcl3):δ7.816-7.828(m,1h),7.707-7.747(m,1h),7.509-7.549(m,1h),7.035-7.075(m,1h),5.103-5.118(m,1h),2.934-2.995(m,1h),2.458-2.510(m,1h)。
[0210]
实例7
[0211]
(4r)-2-(3-溴-2-氟苯基)-4-((四氢-2h-吡喃-2-基)氧基)环戊-2-烯酮
[0212][0213]
用二氯甲烷(100ml)溶解(r)-2-(3-溴-2-氟苯基)-4-羟基环戊-2-烯酮(10.0g,37mmol,来自实例6)。随后向其中添加乙酸(145mg,1.85mmol)和二氢吡啶(4.67g,55.5mmol)。在室温下搅拌反应混合物。藉由tlc监测确认反应完成。用10%nahco
3(水溶液)
(100ml)淬灭反应混合物且对反应混合物进行相分离。收集有机层且将其经无水na2so4干燥。滤出固体且在真空下蒸发掉有机溶剂,得到粗化合物。藉由硅胶层析使用己烷和乙酸乙酯的混合物作为梯度溶离剂来纯化粗产物。标题化合物的产量为10.99g(83%)。
[0214]1h-nmr(400mhz,cdcl3):δ7.873-7.927(m,1h),7.715-7.771(m,1h),7.503-7.524(m,1h),7.016-7.055(m,1h),5.001-5.030(m,1h),4.790-4.868(m,1h),3.853-3.994(m,
4-甲基辛-1-烯-6-炔-1-基)-4-((四氢-2h-吡喃-2-基)氧基)环戊烯酮
[0227][0228]
在-70℃下,向(e)-叔丁基((1-碘-4-甲基辛-1-烯-6-炔-3-基)氧基)二甲基硅烷(130.0g,343.6mmol)于无水二乙醚(1000ml)中的溶液中逐滴添加含1.9m(360ml,684mmol)叔丁基锂的戊烷,且将其在相同温度下搅拌2小时。将碘化铜(65.3g,342.8mmol)和正三丁基膦(180.7g,893.1mmol)于thf(1.3l)中的混合物冷却至-70℃且添加至反应烧瓶中。1小时后,在-70℃下向其中添加2-(3-溴-2-氟苯基)-4-((四氢-2h-吡喃-2-基)氧基)环戊-2-烯酮(91.5g,257.6mmol,来自实例4)于90ml thf中的溶液。用含有氢氧化铵(0.5l)的饱和氯化铵水溶液(4.5l)淬灭反应混合物。对反应混合物进行相分离,且用乙酸乙酯萃取水层。将有机层合并且经无水na2so4干燥。滤出固体且在真空下蒸发有机溶剂,得到426.6g粗标题化合物。
[0229]
实例11
[0230]
(1sr,2sr,3rs,4rs)-2-(3-溴-2-氟苯基)-3-((e)-3-((叔丁基二甲基硅烷基)氧基)-4-甲基辛-1-烯-6-炔-1-基)-4-((四氢-2h-吡喃-2-基)氧基)环戊醇
[0231][0232]
用无水thf(4.3l)稀释(2sr,3rs,4rs)-2-(3-溴-2-氟苯基)-3-((3s,e)-3-((叔丁基二甲基硅烷基)氧基)-4-甲基辛-1-烯-6-炔-1-基)-4-((四氢-2h-吡喃-2-基)氧基)环戊烯酮(426.6g,699.7mmol,来自实例10的粗产物),随后将溶液冷却至-70℃,且接着在-70℃下添加含1.0m l-selectride(258ml,258莫耳)的己烷。添加后,藉由tlc检查反应。混合物藉由过氧化氢(30%水溶液,100ml)淬灭且在0℃下搅拌30分钟。对反应混合物进行相分离,且用乙酸乙酯萃取水层。将有机层合并且经无水na2so4干燥。滤出固体且在真空下蒸发掉有机溶剂。藉由硅胶层析使用己烷和乙酸乙酯的混合物作为梯度溶离剂来纯化粗产物。标题化合物的产量为99.8g(64%,自实例10开始2个步骤)。
[0233]1h-nmr(400mhz,cdcl3):δ7.422(m,1h),7.258-7.338(m,1h),6.991-7.021(m,1h),5.364-5.553(m,2h),4.702-4.747(m,1h),3.854-4.285(m,4h),3.493-3.519(m,1h),3.116-3.221(m,2h),2.395-2.564(m,1h),1.541-2.110(m,14h),0.647-0.910(m,12h),-0.271~-0.026(m,6h)。
[0234]
实例12
[0235]
(((e)-1-((1rs,2rs,3asr,8bsr)-5-溴-2-((四氢-2h-吡喃-2-基)氧基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-1-基)-4-甲基辛-1-烯-6-炔-3-基)氧基)(叔丁基)二甲
基硅烷
[0236][0237]
在60℃下在氮气下,向(1sr,2sr,3rs,4rs)-2-(3-溴-2-氟苯基)-3-((e)-3-((叔丁基二甲基硅烷基)氧基)-4-甲基辛-1-烯-6-炔-1-基)-4-((四氢-2h-吡喃-2-基)氧基)环戊醇(98.3g,166.7mmol,来自实例11)于甲苯(5.45l)和dmpu(115.4g,900.3mmol)中的溶液中,添加t-buok(64.3g,573.1mmol)。藉由tlc监测确认反应完成。混合物用饱和氯化铵水溶液(1000ml)洗涤且经无水na2so4干燥。滤出固体且在真空下蒸发掉有机溶剂。藉由硅胶层析使用己烷和乙酸乙酯的混合物作为梯度溶离剂来纯化粗产物。标题化合物的产量为79.2g(83%)。
[0238]1h-nmr(400mhz,cdcl3):δ7.259(m,1h),7.054(m,1h),6.686-6.714(m,1h),5.521-5.655(m,2h),5.262-5.294(m,1h),4.603-4.662(m,1h),4.049-4.184(m,2h),3.837-3.912(m,1h),3.411-3.689(m,2h),2.441-2.781(m,2h),1.947-2.242(m,3h),1.209-1.785(m,10h),0.833-0.939(m,12h),0.101-0.070(m,6h)。
[0239]
实例13
[0240]
(1rs,2rs,3asr,8bsr)-5-溴-1-((3sr,e)-3-羟基-4-甲基辛-1-烯-6-炔-1-基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-2-醇
[0241][0242]
用乙腈(758ml)稀释(((e)-1-((1rs,2rs,3asr,8bsr)-5-溴-2-((四氢-2h-吡喃-2-基)氧基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-1-基)-4-甲基辛-1-烯-6-炔-3-基)氧基)(叔丁基)二甲基硅烷(75.8g,128.5mmol,来自实例12),和向其中添加3n hcl水溶液(76ml)。在室温下搅拌反应物且藉由tlc检查反应进程。在反应完成后,混合物用10%nahco3水溶液(760ml)中和至ph 7-8,且随后浓缩以移除乙腈。用乙酸乙酯萃取残余物且有机层经无水na2so4干燥。滤出固体且在真空下蒸发掉有机溶剂。藉由硅胶层析使用己烷和乙酸乙酯的混合物作为梯度溶离剂来纯化粗产物。标题11,15-同侧异构物的产量为26.3g(52%)且11,15-反侧异构物(15-差向异构物)的产量为11.1g(22%)。自乙酸乙酯和正己烷的溶剂混合物再结晶化合物,得到结晶化合物(22.83g)。结晶化合物的特征在于在6.7
±
0.2
°
、15.4
±
0.2
°
、19.5
±
0.2
°
、19.9
±
0.2
°
和21.5
±
0.2
°
2θ处具有峰的x射线粉末绕射(xrpd)图。
[0243]1h-nmr(400mhz,cdcl3):δ7.259-7.294(m,1h),6.992-7.032(m,1h),6.698-6.730(m,1h),5.563-5.706(m,2h),5.186-5.235(m,1h),4.029-4.194(m,1h),3.924(m,1h),
叔丁基醚(25ml)进一步洗涤。水层用3n hcl水溶液酸化至ph 3-4且进一步用甲基-叔丁基醚(25ml)萃取。有机层经无水na2so4干燥。滤出固体且在真空下蒸发掉有机溶剂。藉由硅胶层析使用己烷和乙酸乙酯的混合物作为梯度溶离剂来纯化粗产物。标题化合物的产量为1.78g(77%)。
[0253]1h-nmr(400mhz,cdcl3):δ6.892-6.942(m,2h),6.729-6.748(m,1h),5.503-5.656(m,2h),5.038-5.056(m,1h),3.984-4.107(m,1h),3.821-3.893(m,1h),3.214-3.390(m,1h),2.542-2.673(m,3h),2.325-2.394(m,3h),2.044-2.238(m,2h),1.857-1.998(m,3h),1.745-1.788(m,4h),0.979-1.031(d,3h)。
[0254]
13
c-nmr(100mhz,cdcl3):δ178.685,157.204,134.016,133.811,133.325,132.756,129.621,129.576,128.908,123.200,123.169,121.856,121.811,120.551,120.513,84.223,84.132,77.415,77.240,77.225,77.179,76.367,76.079,75.980,58.780,58.735,50.196,50.150,41.103,38.218,38.066,33.209,29.034,24.540,22.453,22.362,15.720,14.847,3.523,3.492。
[0255]
实例16
[0256]
(2s,3r,4r)-2-(3-溴-2-氟苯基)-3-((3s,4s,e)-3-((叔丁基二甲基硅烷基)氧基)-4-甲基辛-1-烯-6-炔-1-基)-4-((四氢-2h-吡喃-2-基)氧基)环戊烯酮
[0257][0258]
2-噻吩基(氰基)铜锂是在干燥氮气下制备。在-10℃下,向噻吩(6.84g,81.4mmol)于无水thf(70ml)中的溶液中逐滴添加含1.6m正丁基锂的己烷(46.3ml,74.1mmol)。在一小时后,使反应混合物冷却至-70℃。溶液经由套管在-70℃下转移至氰化铜(7.3g,81.5mmol)于无水thf(73ml)中的悬浮液中且将其搅拌30分钟。接着,在-70℃下将含正丁基锂的己烷(1.6m,55.5ml,88.8mmol)逐滴添加至叔丁基二甲基(((3s,4s,e)-4-甲基-1-(三丁基锡烷基)辛-1-烯-6-炔-3-基)氧基)硅烷(40.1g,74mmol)的溶液中,持续30分钟。在-70℃下经由套管向反应混合物中添加2-噻吩基(氰基)铜锂且搅拌30分钟。随后,向其中添加(4r)-2-(3-溴-2-氟苯基)-4-((四氢-2h-吡喃-2-基)氧基)环戊-2-烯酮(13.2g,37.1mmol,来自实例7)的溶液。用含有氢氧化铵(60ml)的饱和氯化铵水溶液(540ml)淬灭反应混合物。对反应混合物进行相分离,且用乙酸乙酯萃取水层。将有机层合并且经无水na2so4干燥。滤出固体且在真空下蒸发有机溶剂,得到60.2g粗标题化合物。
[0259]
实例17
[0260]
(1s,2s,3r,4r)-2-(3-溴-2-氟苯基)-3-((3s,4s,e)-3-((叔丁基二甲基硅烷基)氧基)-4-甲基辛-1-烯-6-炔-1-基)-4-((四氢-2h-吡喃-2-基)氧基)环戊醇
[0261][0262]
藉由遵循实例11的步骤,自(2s,3r,4r)-2-(3-溴-2-氟苯基)-3-((3s,4s,e)-3-((叔丁基二甲基硅烷基)氧基)-4-甲基辛-1-烯-6-炔-1-基)-4-((四氢-2h-吡喃-2-基)氧基)环戊烯酮(60.2g,来自实例16)获得化合物。标题化合物的产量为13.8g(65%,自实例16开始2个步骤)。
[0263]1h-nmr(400mhz,cdcl3):δ7.422(m,1h),7.289-7.352(m,1h),7.001(m,1h),5.359-5.542(m,2h),4.699-4.743(m,1h),3.850-4.346(m,4h),3.497-3.513(m,1h),3.130-3.265(m,2h),2.378-2.578(m,1h),1.524-2.026(m,14h),0.642-0.955(m,12h),-0.182-0.089(m,6h)。
[0264]
实例18
[0265]
(((3s,4s,e)-1-((1r,2r,3as,8bs)-5-溴-2-((四氢-2h-吡喃-2-基)氧基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-1-基)-4-甲基辛-1-烯-6-炔-3-基)氧基)(叔丁基)二甲基硅烷
[0266][0267]
藉由遵循实例12的步骤,自(1s,2s,3r,4r)-2-(3-溴-2-氟苯基)-3-((3s,4s,e)-3-((叔丁基二甲基硅烷基)氧基)-4-甲基辛-1-烯-6-炔-1-基)-4-((四氢-2h-吡喃-2-基)氧基)环戊醇(13.8g,22.6mmol,来自实例17)获得化合物。标题化合物的产量为10.9g(81%)。
[0268]1h-nmr(400mhz,cdcl3):δ7.259(m,1h),7.014-7.055(m,1h),6.670-6.689(m,1h),5.481-5.654(m,2h),5.242-5.264(m,1h),4.603-4.664(m,1h),4.048-4.077(m,2h),3.818-3.912(m,1h),3.414-3.713(m,2h),2.408-2.798(m,2h),2.057-2.233(m,3h),1.259-1.787(m,10h),0.856-0.950(m,12h),-0.007-0.095(m,6h)。
[0269]
实例19
[0270]
(1r,2r,3as,8bs)-5-溴-1-((3s,4s,e)-3-羟基-4-甲基辛-1-烯-6-炔-1-基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-2-醇
[0271][0272]
藉由遵循实例13的步骤,自(((3s,4s,e)-1-((1r,2r,3as,8bs)-5-溴-2-((四氢-2h-吡喃-2-基)氧基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-1-基)-4-甲基辛-1-烯-6-炔-3-基)氧基)(叔丁基)二甲基硅烷(9.9g,16.8mmol,来自实例18)获得化合物。标题化合物的产量为5.55g(84%)。再结晶后获得结晶化合物(4.12g,75%)。结晶化合物的特征在于在6.7
±
0.2
°
、15.4
±
0.2
°
、19.5
±
0.2
°
、19.9
±
0.2
°
和21.5
±
0.2
°
2θ处具有峰的x射线粉末绕射图。
[0273]1h-nmr(400mhz,cdcl3):δ7.258-7.277(m,1h),6.967-6.985(m,1h),6.681-6.720(m,1h),5.523-5.655(m,2h),5.158-5.213(m,1h),3.989-4.024(m,1h),3.838-3.884(m,1h),3.463-3.508(m,1h),3.285-3.296(m,1h),2.949(s,1h),2.665-2.733(m,1h),2.343-2.407(m,1h),2.221-2.242(m,2h),1.972-2.044(m,1h),1.715-1.853(m,4h),0.961-0.977(m,3h)。
[0274]
实例20
[0275]
4-((1r,2r,3as,8bs)-2-羟基-1-((3s,4s,e)-3-羟基-4-甲基辛-1-烯-6-炔-1-基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-5-基)丁酸甲酯
[0276][0277]
藉由遵循实例14的步骤,自(1r,2r,3as,8bs)-5-溴-1-((3s,4s,e)-3-羟基-4-甲基辛-1-烯-6-炔-1-基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-2-醇(2.0g,5.1mmol,来自实例19)获得化合物。标题化合物的产量为1.47g(70%)。再结晶后获得结晶化合物(1.11g,75%)。结晶化合物的特征在于在8.2
±
0.2
°
、10.8
±
0.2
°
、18.5
±
0.2
°
、20.9
±
0.2
°
和22.1
±
0.2
°
2θ处具有峰的x射线粉末绕射图。
[0278]1h-nmr(400mhz,cdcl3):δ6.886-6.931(m,2h),6.711-6.748(t,1h),5.501-5.656(m,2h),5.015-5.070(m,1h),3.969-4.007(t,1h),3.809-3.869(m,1h),3.628(s,3h),3.541(s,1h),3.341-3.386(t,1h),3.168(s,1h),2.564-2.677(m,3h),2.291-2.373(m,3h),2.168-2.220(m,2h),1.876-1.970(m,3h),1.696-1.778(m,4h),0.955-0.971(m,3h)。
[0279]
13
c-nmr(100mhz,cdcl3):δ174.173,157.194,134.083,133.324,129.582,128.830,123.290,121.749,120.428,84.042,77.279,77.150,76.285,75.882,58.843,51.458,50.129,41.211,38.175,33.416,29.173,24.710,22.358,15.694,3.474。
[0280]
实例21
[0281]
4-((1r,2r,3as,8bs)-2-羟基-1-((3s,4s,e)-3-羟基-4-甲基辛-1-烯-6-炔-1-基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-5-基)丁酸(贝前列素-314d)
[0282][0283]
藉由遵循实例15的步骤,自4-((1r,2r,3as,8bs)-2-羟基-1-((3s,4s,e)-3-羟基-4-甲基辛-1-烯-6-炔-1-基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-5-基)丁酸甲酯(406mg,0.98mmol,来自实例20)获得化合物。标题化合物的产量为353mg(90%)。再结晶后获得结晶化合物(251g,71%)。结晶化合物的特征在于在6.1
±
0.2
°
、6.6
±
0.2
°
、7.2
±
0.2
°
、12.1
±
0.2
°
和16.3
±
0.2
°
2θ处具有峰的x射线粉末绕射图。
[0284]1h-nmr(400mhz,cdcl3):δ6.904-6.941(m,2h),6.722-6.759(t,1h),5.522-5.672(m,2h),5.032-5.086(m,1h),3.996-4.033(m,1h),3.850-3.909(m,1h),3.369-3.412(m,1h),2.536-2.668(m,3h),2.301-2.416(m,3h),2.229(m,2h),1.879-2.005(m,3h),1.717-1.788(m,4h),0.967-0.984(d,3h)。
[0285]
13
c-nmr(100mhz,cdcl3):δ178.287,157.194,134.052,133.293,129.582,128.937,123.199,121.810,120.504,84.148,77.233,77.218,76.353,75.966,58.752,50.137,41.082,38.061,33.135,29.022,24.543,22.342,15.716,3.519。
[0286]
实例22
[0287]
4-((1r,2r,3as,8bs)-2-((叔丁基二甲基硅烷基)氧基)-1-((3s,4s,e)-3-((叔丁基二甲基硅烷基)氧基)-4-甲基辛-1-烯-6-炔-1-基)-2,3,3a,8b-四氢-1h-环戊并[b]苯并呋喃-5-基)丁酸甲酯
[0288][0289]
化合物自(r)-4-(3-(3-((叔丁基二甲基硅烷基)氧基-5-侧氧基环-1-烯-1-基)-2-氟苯基)丁酸甲酯(10.0g,24.6mmol,来自实例9)和叔丁基二甲基(((3s,4s,e)-4-甲基-1-(三丁基锡烷基)辛-1-烯-6-炔-3-基)氧基)硅烷(39.96g,73.8mmol)获得,接着进行实例10、11和12的相同步骤。标题化合物的产量为7.6g(48%,3个步骤)。
[0290]1h-nmr(400mhz,cdcl3):δ6.963-6.982(d,1h),6.895-6.912(d,1h),6.708-6.745(t,1h,j=7.6hz),5.496-5.623(m,2h),5.073-5.126(m,1h),4.019-4.048(m,1h),3.902-3.952(m,1h),3.652(s,3h),3.433-3.473(m,1h),2.525-2.599(m,3h),2.417-2.484(m,1h),2.319-2.356(m,2h),2.192-2.252(m,1h),2.060-2.120(m,1h),1.891-2.012(m,3h),
1.780-1.792(m,3h),1.628-1.745(m,1h),0.758-0.941(m,21h),-0.049-0.091(m,12h)。
[0291]
13
c-nmr(100mhz,cdcl3):δ174.105,157.331,132.861,131.350,130.212,128.405,122.956,121.901,120.109,84.801,77.909,77.142,76.421,75.958,58.114,51.412,49.962,42.380,39.746,33.568,29.302,29.234,25.887,25.674,25.629,24.756,21.940,18.168,17.819,15.542,3.458,-2.970,-3.980,-4.678,-4.875。
[0292]
虽然已参考说明性实例描述了本发明,但应理解,所属领域的技术人员可易于实现的任何修改或更改将属于本说明书和所附权利要求书的揭示内容的范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。