1.本发明涉及一种包含磺酰胺基和含氮杂环化合物的、在中高温(120-180℃)和低加湿条件下(湿度小于等于30rh%)也具有优异质子导电性的聚合物,使用该聚合物可以实现优异的长期耐久性的燃料电池用聚合物电解质膜,固体高分子型燃料电池。
背景技术:
2.燃料电池是通过将电化学反应氧化燃料,例如:氢或甲醇,从而产生电能的发电器。特别是固体高分子型燃料电池,具有较高的能量密度,因此预期可被广泛应用于小规模的分散型发电设施以及汽车、船舶等移动体的发电装置。燃料电极(阳极)、氧电极(阴极)和安置在两电极间的聚合物电解质膜的结构称为膜电极组件(简称mea)。再将双极板与膜电极组件交替叠合,之间嵌入密封件,经前后端板压紧拴牢,即构成固体高分子型燃料电池。
3.在固体高分子型燃料电池中,阳极进行的反应如式(1)所示:
4.h2→
2h
2e-ꢀꢀꢀꢀꢀ
(1)
5.式(1)产生的电子通过外部电路,通过外部负荷后到达阴极侧。产生的质子通过电渗透从聚合物电解质膜内由阳极侧移动到阴极侧。
6.另一方面,在阴极进行如式(2)所示的反应:
7.4h
o2 4e-→
2h2o
ꢀꢀꢀ
(2)
8.高的质子导电率是作为聚合物电解质膜的首要特性。另外,考虑到长期耐久性,需要耐受燃料电池工作时溶胀
·
干燥的反复过程中的机械强度和物理耐久性,要求聚合物电解质膜在吸水溶胀时具有高的膜平行方向尺寸稳定性。另外作为要求的其他特性,可以列举出燃料阻隔性,耐强氧化气氛的化学稳定性等等。
9.固体高分子型燃料电池的工作温度,在高温下,如中高温(120-180℃),能够简化燃料电池系统的水热管理,提高电化学反应动力学,避免催化剂被co中毒等等优点而被广泛关注。在现行的商业化应用的聚合物电解质膜中,广泛使用全氟磺酸系聚合物,其代表商品为nafion(注册商标)(杜邦公司制)。但是全氟磺酸系聚合物具有价格昂贵,合成污染严重等问题。且全氟磺酸系聚合物的热变形温度较低,为避免高温时蠕变降低耐久性,通常的使用温度须保持在80℃以下。
10.为了解决全氟磺酸系聚合物的问题,目前大量研究了不含氟原子,在主链芳香环上接枝磺酸基的芳香族聚合物,然而这些含磺酸基的聚合物为了提高质子导电率,要求磺酸基的量越高越好。但是,磺酸基的吸水性大,常常导致聚合物电解质膜吸水溶胀时的尺寸稳定性,从而限制磺酸基的导入量。且温度高于100℃时,由于磺酸基的结合水降低,也存在中高温下质子导电性不足的情况。
11.含氮杂环化合物具有原生的质子导电性,其质子导电率一定程度上不依赖环境湿度,因此在中高温质子导电性聚合物的开发中被认为具有很好的应用前景。专利文献1通过含氮杂环的二胺单体,在磺化聚酰亚胺主链中引入氮杂环,然后与磷酸掺杂制备聚酰亚胺聚合物电解质膜;专利文献2将三氮唑盐引入磺化聚醚砜的侧链制备聚合物电解质膜;专利
文献3合成了聚苯乙烯-1,2,3-三氮唑聚合物,将其与聚苯乙烯膦酸聚合物复合制备聚合物电解质膜;专利文献4将三氮唑和膦酸基团同时引入聚醚砜的侧链,制备聚合物电解质膜。
12.这些专利文献都在一定程度上提高了聚合物电解质膜在低加湿或无水条件下的质子导电率,但是仍不足够,如何进一步发挥出含氮杂环的在中高温时的质子导电率,同时实现聚合物电解质膜的吸水溶胀时的尺寸稳定性从而获得长期耐久性,仍然需要进一步的研究。
13.现有技术文献
14.专利文献
15.专利文献1:cn103694490a
16.专利文献2:cn104927043a
17.专利文献3:cn104610674a
18.专利文献4:cn107033350a
技术实现要素:
19.所要解决的技术课题
20.本发明的课题提供一种包含磺酰胺基和含氮杂环化合物的质子导电聚合物,在中高温(120-180℃)和低加湿条件下(湿度小于等于30rh%)具有优异质子导电性;在制备成聚合物电解质膜时,同时具有高的吸水溶胀后的尺寸稳定性;在使用到固体高分子型燃料电池中时,具有高输出和长期耐久性。此外,本发明提供含磺酰胺基和含氮杂环化合物的质子导电聚合物的制备方法。
21.用于解决课题的技术手段
22.本发明人等反复进行了深入研究,结果发现,通过在质子导电聚合物中包含聚合物骨架、与所述聚合物骨架直接连接的磺酰胺基、和所述磺酰胺基通过共价键连接的含氮杂环化合物,能够解决上述课题。
23.含氮杂环化合物通常表现出质子供体和受体性子,同时利用其自解离度高的特性,提供质子除了水合质子和氢键网络之外额外的跳跃位点,从而提高质子导电聚合物的低加湿条件时的质子导电性。然而并不是所有的含氮杂环化合物都表现出很好的质子导电性。从提高质子导电性出发,本发明通过将磺酰胺基与含氮杂环化合物通过共价键连接,一方面,磺酰胺基本身可以和水合质子结合,利用vehicle机理在磺酰胺基之间进行质子扩散;另一方面,又能与含氮杂环化合物进行相互作用构建质子跳跃网络,利用grotthuss机理,质子跳跃到可以结合和解离质子的邻近含氮杂环基团上,帮助质子扩散。进一步的,磺酰胺基的吸电子能力,能降低含氮杂环化合物的pka值,促进含氮杂环化合物的与质子结合和解离的能力,从而使得质子导电聚合物降低对自由水的依赖,即表现出在低价湿条件下优异的质子导电率。
24.从更进一步提高质子导电聚合物的质子导电性出发,连接含氮杂环化合物的磺酰胺基具有如结构式(s1)所示的结构,其中r1为含氮杂环化合物,*为式(s1)所示结构与所述聚合物骨架的结合位点。当磺酰胺基通过仲胺键链接含氮杂环化合物时,含氮杂环化合物的pka值进一步减弱,即当含氮杂环化合物的质子被脱去后,氮原子上的孤对电子更加容易再结合质子,从而进一步提高低加湿下的质子导电率。
[0025][0026]
本发明的质子导电聚合物,从质子导电性出发,进一步优选与磺酰胺基通过共价键连接的含氮杂环化合物的杂环中同时含有n-h键和具有未与h结合的n原子。呈酸性的n-h键,容易断裂解离出质子;未与h结合的n原子,具有孤对电子,易与质子结合。当他们同时存在于杂环中时,同时具有和质子的结合和解离的能力。
[0027]
本发明的质子导电聚合物,更优选所述含氮杂环化合物为五元含氮杂环化合物。这是由于五元含氮杂环化合物具有二维平面的五元环共轭体系结构,呈酸性的n-h键,更容易断裂解离出质子;未与h结合的n原子上的孤对电子更易与质子结合。
[0028]
作为本发明的五元含氮杂环化合物,可以列举出1,2-吡唑、1,3-咪唑、1,2,3-三氮唑、1,2,4-三氮唑、1,2,3,4-四氮唑,但不限于这些。杂环中可以含有其他杂原子,如硫、氧等。含氮杂环化合物的杂环上还可以包含其他取代基。但从成本,以及更多的n-h键或含孤对电子的n原子将具有更高的质子导电性出发,更优选含氮杂环化合物为1,2,3-三氮唑、1,2,4-三氮唑、1,2,3,4-四氮唑中的一种。
[0029]
作为本发明聚合物骨架,没有特别限制,只要是可兼具机械性能和化学稳定性的聚合物结构即可。从提高化学稳定性和原料成本考虑,优选全氟系聚合物骨架或含芳香环的碳氢系聚合物骨架。但更优选耐热性和气体阻隔性高的含芳香环的碳氢系聚合物骨架。芳香环不仅可以包含烃系芳香环,也可以包含杂芳环等。具体的例子可以举出聚砜、聚醚砜、聚醚酮、聚苯醚、聚芳撑醚系聚合物、聚芳硫醚、聚苯硫醚、聚苯硫醚砜、聚对苯撑、聚芳撑、聚芳酮、聚芳撑氧化膦、聚苯并唑、聚苯并咪唑、聚苯并噻唑、芳香族聚酰胺、聚酰亚胺、聚醚酰亚胺、或聚酰亚胺砜。
[0030]
本发明的质子导电性聚合物可形成质子导电通道,从增加质子导电通道区域的磺酰胺基和含氮杂环化合物的密度,更有利于提高质子导电性出发,更优选本发明的质子导电聚合物具有结构式(s2)所示的组成单元,
[0031][0032]
其中,a表示直接键合、醚键、羰基或砜基;b各自独立地表示直接键合、醚键、—s—、—nh—co—、—n—(co)2—;r1表示含氮杂环化合物;n1和n2表示1或2的整数。考虑到化学稳定性和成本,a优选为醚键、羰基或砜基;b优选为醚键、—s—、—n—(co)2—。考虑到物理耐久性,a更优选醚键或羰基;b更优选为醚键和—n—(co)2—。
[0033]
从确保质子导电通道的连续性和改良聚合物吸水溶胀导致的尺寸稳定性的角度考虑,作为本发明的质子导电聚合物为嵌段共聚物,包含实质不含离子性基团的聚合物链段。本发明中,链段是指嵌段共聚物中的部分结构,表示由一种重复单元或多种重复单元的
组合构成的,数均分子量2000以上的部分。实质不含离子性基团指的是链段中包含磺酸基、磺酰胺基的组成单元在所有该链段的组成单元的摩尔比例在5%以下。
[0034]
本发明所获得的嵌段共聚物是将两种或两种以上的互相不兼容链段即含磺酰胺基与不含磺酰胺基的链段形成一个聚合物链。该嵌段共聚物中,由于化学相同结构的链段相互吸引,不同的结构的链段相互排斥,而形成相分离,形成含各链段链的微域,通过包含各链段的微域聚集形成的高度有序的纳米或微米相分离结构。而当含磺酰胺基和含氮杂环化合物的链段形成连续的相分离结构通道时,将获得更优秀的质子导电性。而不含磺酰胺基的链段形成的连续相分离结构又为聚合物提供物理支撑,实现优秀的尺寸稳定性。
[0035]
本发明所述的质子导电聚合物的制备方法中,通过含磺酰胺基和通过共价键连接的含氮杂环化合物的芳香族的化合物与其他化合物通过缩合聚合得到。
[0036]
作为含磺酰胺基和通过共价键连接的含氮杂环化合物的芳香族的化合物为具有可反应端基的二氨基化合物、二卤素基化合物。考虑到反应性,卤素可以优选为氟或氯。具体的可以列举出式(s3-1)至(s3-14)所示结构的化合物,但本发明不限于此。
[0037]
[0038][0039]
式(s3-1)至(s3-14)所示结构中的s1代表式(s1)所示的结构,式(s1)中r1为含氮杂环化合物,*为式(s1)所示结构与所述聚合物骨架的结合位点。
[0040][0041]
作为与上述含磺酰胺基和通过共价键连接的含氮杂环化合物的芳香族的化合物进行缩聚的其他化合物,可以列举出与其具有化学聚合活性的二羟基酚化合物、二羧酸基羧酸化合物、二羧酸酐化合物等。
[0042]
作为本发明的质子导电聚合物的制备方法,可以由上述含磺酰胺基和通过共价键连接的含氮杂环化合物的芳香族的二氨基化合物、二卤素基化合物,与二羟基酚化合物、二羧酸基酸化合物、二羧酸酐化合物以任意比例缩合聚合得到,只要聚合物能实现达到可用的机械性能即可。在聚合过程中,也可以添加一定比例的不含取代基团的二氨基化合物、二卤素基化合物进行缩合聚合。但从质子导电性出发,含磺酰胺基和通过共价键连接的含氮杂环化合物的芳香族的二氨基化合物、二卤素基化合物与不含取代基团的二氨基化合物、二卤素基化合物的摩尔比不小于0.5。
[0043]
作为本发明的质子导电聚合物的制备方法,为了合成嵌段共聚物,优选将上述含磺酰胺基和通过共价键连接的含氮杂环化合物的芳香族的二氨基化合物、二卤素基化合物和二羟基酚化合物、二羧酸基酸化合物、二羧酸酐化合物合成预聚物。再与不含取代基团的二氨基化合物、二卤素基化合物和二羟基酚化合物、二羧酸基酸化合物、二羧酸酐化合物合成的预聚物进行缩合聚合,得到嵌段共聚物。
[0044]
本发明也可以提供一种聚合物电解质膜,其通过使用本发明的质子导电聚合物而
得。具体而言,本发明的质子导电聚合物可以用作燃料电池中的聚合物电解质膜,该电解质膜可以位于燃料电池的阳极和阴极之间,用于传递质子。
[0045]
本发明还提供一种使用了本发明的聚合物电解质膜的固体高分子型燃料电池。
附图说明
[0046]
图1是实施例1~8、比较例1~3的质子导电聚合物电解质膜90%湿度时的质子导电率。
[0047]
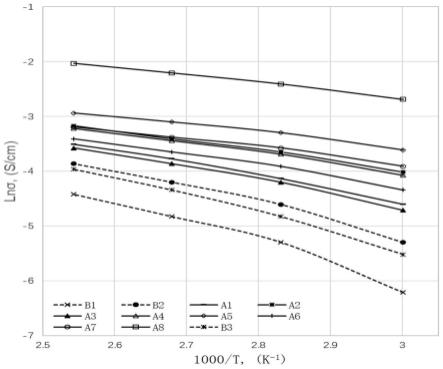
图2是实施例1~8、比较例1~3的质子导电率和温度的arrhenius曲线。
具体实施方式
[0048]
下面给出实施例对本发明作进一步说明,在此提供这些实施例的目的仅在于对本发明进行说明,而不在于对本发明的范围进行限定。此外,测量各种性质的条件如下:
[0049]
(1)质子导电率(σ)
[0050]
将聚合物电解质膜样品浸渍于25℃纯水中24小时,然后在60℃,相对湿度90rh%的恒温恒湿槽中保持30分钟。使用由solartron公司制造的电化学测量系统(solartron 1287electrochemical interface及solartron 1255b frequency response analyzxer)作为测量装置,通过二端子法进行固定电位阻抗测量,获得质子导电率。交流振幅设为50毫伏。测试样品为10毫米宽、50毫米长的薄膜。测量夹具由酚醛树脂制造,测量的部分是松开的。电极使用铂板(厚100微米,2片板)。将电极以10毫米的电极间距放置在样品薄膜的前后侧,使彼此平行并与样品薄膜的纵向正交。
[0051]
保持相对湿度90%不变,将恒温恒湿槽湿槽温度至80℃,100℃,120℃后各保持30分钟,测试各温度下的质子导电率。保持120℃不变,将恒温恒湿槽的相对湿度降低到30rh%后,继续保持30分钟,测试120℃,30%rh的质子导电率。
[0052]
利用90rh%湿度下的质子导电率的自然对数和开尔文温度的倒数可以画出质子导电率随温度变化的arrhenius曲线,根据曲线的斜率k,可以计算出质子导电活化能ea=-k
×
r,r为摩尔气体常数。质子导电活化能的值越低,表明质子导电聚合物更具有通过grotthuss机理进行质子传导的潜力,降低了对自由水的依赖性,在中高温低加湿的情况下具有更好的质子导电能力。
[0053]
(2)热水溶胀度
[0054]
聚合物电解质膜的尺寸稳定性通过在80℃热水中测量尺寸变化率所得的热水溶胀度进行评价。将聚合物电解质膜切成5公分宽的正方形,在25℃,55%湿度的房间中静置一夜,测量长(l1)和宽(h1),在25℃水中浸渍24小时,再在80℃热水中浸渍2小时再迅速以游标卡尺测量湿膜的长(l2)和宽(h2)。通过(l2-l1)/l1*100%和(h2-h1)/h1*100%计算的数值的平均值为热水溶胀度。
[0055]
以下合成例,实施例中所用的到各试剂的品名、纯度、购买渠道如表1所示:
[0056]
表1试剂的品名、纯度、购买渠道
[0057]
品名简称纯度购买自1,4,5,8-萘四甲酸二酐ntda98%阿拉丁试剂有限公司4,4
’‑
二氨基二苯醚oda99%梯希爱试剂有限公司
二氟二苯甲酮dfbp99%梯希爱试剂有限公司二羟基二苯甲酮ddbp99%梯希爱试剂有限公司二氟二苯砜bfs99%梯希爱试剂有限公司乙醇钾 95%aldrich2,2
’‑
苯甲酸ba99%梯希爱试剂有限公司2,5-双(4-氨苯基)-1,3,4-恶二唑baoz98%阿拉丁试剂有限公司3-氨基吡唑 98%阿拉丁试剂有限公司3-氨基-1,2,4-三氮唑 98%梯希爱试剂有限公司4-氨基-1,2,4-三氮唑 98%梯希爱试剂有限公司5-氨基-1h-四氮唑 98%梯希爱试剂有限公司碘 ar国药试剂二异丙基氨基锂溶液 2m国药试剂五氯化磷 98%国药试剂三氯甲烷 ar国药试剂二氯甲烷 ar国药试剂四氢呋喃thfar国药试剂盐酸 ar国药试剂浓硫酸 98%国药试剂氢氧化钠 ar国药试剂间甲酚 ar国药试剂异丙醇ipaar国药试剂丙酮acear国药试剂三乙胺teaar国药试剂甲苯 ar国药试剂碳酸钾 ar国药试剂n-甲基吡咯烷酮nmpar国药试剂
[0058]
二磺酸化的4,4
’‑
二氨基二苯醚(odads)按照专利cn102838764a中记载的方法进行磺化制备;二磺酸化二氟二苯砜(bfsds)和四磺酸化二氟二苯甲酮(dfbpts)按照专利cn103814062a的磺化方法进行制备。
[0059]
(合成例1)
[0060]
如结构式(s4-1)所示的含磺酰胺基和3-氨基吡唑的化合物(odadspz)
[0061][0062]
将odads(20g)、五氯化磷(26.4g)以及三氯甲烷(140ml)先后分别加入到配有机械
搅拌和冷凝管的三口烧瓶中,升温至70℃,反应3h;反应后,将反应生成物倒入冰水中,三氯甲烷有机相和水相分层,取水相旋干,而后60℃真空烘箱烘12h,得中间产物。将中间产物配制成20wt%的水溶液。
[0063]
将3-氨基吡唑(4.2g)和10%的氢氧化钠溶液(20.0ml)先后分别加入到配有机械搅拌和冷凝管的三口烧瓶中,反应30min后,升温至40℃,缓慢滴加(3h)步骤(1)生成的中间产物水溶液(49.9g)。滴加完毕后,用20ml的10%稀盐酸溶液调整ph值至5-6,用三氯甲烷洗涤后,取水相用大量ipa析出固体,烘箱120℃减压干燥得到式(s4-1)所示的化合物odadspz。
[0064]
(合成例2)
[0065]
如结构式(s4-2)所示的含磺酰胺基和3-氨基-1,2,4-三氮唑的化合物(odads3stz)
[0066][0067]
将odads(20g)、五氯化磷(26.4g)以及三氯甲烷(40ml)先后分别加入到配有机械搅拌和冷凝管的三口烧瓶中,升温至70℃,反应3h;反应后,将反应生成物倒入冰水中,三氯甲烷有机相和水相分层,取水相旋干,而后60℃真空烘箱烘12h,得中间产物。将中间产物配制成20wt%的水溶液。
[0068]
将3-氨基-1,2,4-三氮唑(4.2g)和10%的氢氧化钠溶液(20.0ml)先后分别加入到配有机械搅拌和冷凝管的三口烧瓶中,反应30min后,升温至40℃,缓慢滴加(3h)步骤(1)生成的中间产物水溶液(49.9g)。滴加完毕后,用20ml的10%稀盐酸溶液调整ph值至5-6,用三氯甲烷洗涤后,取水相用大量ipa析出固体,烘箱120℃减压干燥得到式(s4-2)所示的化合物odads3stz。
[0069]
(合成例3)
[0070]
如结构式(s4-3)所示的含磺酰胺基和4-氨基-1,2,4-三氮唑的化合物(odads4stz)
[0071][0072]
将odads(20g)、五氯化磷(26.4g)以及三氯甲烷(40ml)先后分别加入到配有机械
搅拌和冷凝管的三口烧瓶中,升温至70℃,反应3h;反应后,将反应生成物倒入冰水中,三氯甲烷有机相和水相分层,取水相旋干,而后60℃真空烘箱烘12h,得中间产物。将中间产物配制成20wt%的水溶液。
[0073]
将4-氨基-1,2,4-三氮唑(4.2g)和10%的氢氧化钠溶液(20.0ml)先后分别加入到配有机械搅拌和冷凝管的三口烧瓶中,反应30min后,升温至40℃,缓慢滴加(3h)步骤(1)生成的中间产物水溶液(49.9g)。滴加完毕后,用20ml的10%稀盐酸溶液调整ph值至5-6,用三氯甲烷洗涤后,取水相用大量ipa析出固体,烘箱120℃减压干燥得到式(s4-3)所示的化合物odads4stz。
[0074]
(合成例4)
[0075]
如结构式(s4-4)所示的含磺酰胺基和5-氨基-1h-四氮唑的化合物(odads5atz)
[0076][0077]
将odads(20g)、五氯化磷(26.4g)以及三氯甲烷(40ml)先后分别加入到配有机械搅拌和冷凝管的三口烧瓶中,升温至70℃,反应3h;反应后,将反应生成物倒入冰水中,三氯甲烷有机相和水相分层,取水相旋干,而后60℃真空烘箱烘12h,得中间产物。将中间产物配制成20wt%的水溶液。
[0078]
将5-氨基-1h-四氮唑(4.3g)和10%的氢氧化钠溶液(20.0ml)先后分别加入到配有机械搅拌和冷凝管的三口烧瓶中,反应30min后,升温至40℃,缓慢滴加(3h)步骤(1)生成的中间产物水溶液(49.9g)。滴加完毕后,用20ml的10%稀盐酸溶液调整ph值至5-6,用三氯甲烷洗涤后,取水相用大量ipa析出固体,烘箱120℃减压干燥得到式(s4-4)所示的化合物odads5atz
[0079]
(合成例5)
[0080]
如结构式(s4-5)所示的含磺酰胺基和3-氨基-1,2,4-三氮唑的化合物(bfsds3stz)
[0081][0082]
将bfsds(17g)、五氯化磷(26.4g)以及三氯甲烷(40ml)先后分别加入到配有机械
搅拌和冷凝管的三口烧瓶中,升温至70℃,反应3h;反应后,将反应生成物倒入冰水中,三氯甲烷有机相和水相分层,取水相旋干,而后60℃真空烘箱烘12h,得中间产物。将中间产物配制成20wt%的水溶液。
[0083]
将3-氨基-1,2,4-三氮唑(4.2g)和10%的氢氧化钠溶液(20.0ml)先后分别加入到配有机械搅拌和冷凝管的三口烧瓶中,反应30min后,升温至40℃,缓慢滴加(3h)步骤(1)生成的中间产物水溶液(51.3g)。滴加完毕后,用20ml的10%稀盐酸溶液调整ph值至5-6,用三氯甲烷洗涤后,取水相用大量ipa析出固体,烘箱120℃减压干燥得到式(s4-5)所示的化合物bfsds3stz
[0084]
(合成例6)
[0085]
如结构式(s4-6)所示的含磺酰胺基和3-氨基-1,2,4-三氮唑的化合物(dfbpts3stz)
[0086][0087]
将dfbpts(20.8g)、五氯化磷(26.4g)以及三氯甲烷(40ml)先后分别加入到配有机械搅拌和冷凝管的三口烧瓶中,升温至70℃,反应3h;反应后,将反应生成物倒入冰水中,三氯甲烷有机相和水相分层,取水相旋干,而后60℃真空烘箱烘12h,得中间产物。将中间产物配制成20wt%的水溶液。
[0088]
将3-氨基-1,2,4-三氮唑(4.2g)和10%的氢氧化钠溶液(20.0ml)先后分别加入到配有机械搅拌和冷凝管的三口烧瓶中,反应30min后,升温至40℃,缓慢滴加(3h)步骤(1)生成的中间产物水溶液(84.5g)。滴加完毕后,用20ml的10%稀盐酸溶液调整ph值至5-6,用三氯甲烷洗涤后,取水相用大量ipa析出固体,烘箱120℃减压干燥得到式(s4-6)所示的化合物dfbpts3stz
[0089]
(合成例7)
[0090]
如结构式(s4-7)所示的含磺酰基和1,2,4-三氮唑的化合物(bfpstzt)
[0091][0092]
将bfs(5.0g,22.9mmol,1个当量)溶解在60ml无水四氢呋喃中,将温度降低到-78
℃,加二异丙基氨基锂溶液(45.8ml,2mol/l,4个当量)与反应液中,滴加完毕,继续反应1小时,然后一次性加入碘单质(34.9g,137.4mmol,5个当量)的四氢呋喃溶液,完毕后,于室温继续搅拌12小时,加入亚硫酸钠除去多余的碘,然后导入水中,二氯甲烷萃取,氯化钠饱和水溶液洗,并用异丙醇/二氯甲烷混合溶剂重结晶得到碘取代的dfbp中间产物。
[0093]
取上述碘取代的dfbp中间产物(10g,13.8mmol,1个当量)溶解于60ml二氯甲烷中,将3-巯基-1,2,4-三氮唑(2.8g,27.6mmol,2个当量)和乙醇钾的二氯甲烷溶液滴加到上述溶液中,室温下搅拌24个小时,然后加入3氯过氧苯甲酸和20%的乙醇溶液进行氧化,并用大量异丙醇沉淀,烘箱120℃减压干燥得到式(s4-7)所示的化合物bfpstzt。
[0094]
实施例1
[0095]
odadspz(8.74g,16.7mmol)、oda(3.34g,16.7mmol)在室温、氮气气氛下溶于间甲酚(150ml)、三乙胺(8.43g,83.3mmol)混和溶剂中,向反应瓶中加入ntda(9.20g,34.3mmol)及苯甲酸(6.10g,50mmol),将反应体系加热至80℃,并在此温度下反应4小时,然后再升至180℃,并反应16小时,反应液用丙酮沉淀,得到质子导电聚合物a1。其结构式如表2所示。
[0096]
将上述质子导电聚合物a1(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0097]
实施例2
[0098]
odads3stz(8.77g,16.7mmol)、oda(3.34g,16.7mmol)在室温、氮气气氛下溶于间甲酚(150ml)、三乙胺(8.43g,83.3mmol)混和溶剂中,向反应瓶中加入ntda(9.20g,34.3mmol)及苯甲酸(6.10g,50mmol),将反应体系加热至80℃,并在此温度下反应4小时,然后再升至180℃,并反应16小时,反应液用丙酮沉淀,得到质子导电聚合物a2。其结构式如表2所示。
[0099]
将上述质子导电聚合物a2(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0100]
实施例3
[0101]
odads4stz(8.77g,16.7mmol)、oda(3.34g,16.7mmol)在室温、氮气气氛下溶于间甲酚(150ml)、三乙胺(8.43g,83.3mmol)混和溶剂中,向反应瓶中加入ntda(9.20g,34.3mmol)及苯甲酸(6.10g,50mmol),将反应体系加热至80℃,并在此温度下反应4小时,然后再升至180℃,并反应16小时,反应液用丙酮沉淀,得到质子导电聚合物a3。其结构式如表2所示。
[0102]
将上述质子导电聚合物a3(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留
的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0103]
实施例4
[0104]
odads5atz(8.79g,16.7mmol)、oda(3.34g,16.7mmol)在室温、氮气气氛下溶于间甲酚(150ml)、三乙胺(8.43g,83.3mmol)混和溶剂中,向反应瓶中加入ntda(9.20g,34.3mmol)及苯甲酸(6.10g,50mmol),将反应体系加热至80℃,并在此温度下反应4小时,然后再升至180℃,并反应16小时,反应液用丙酮沉淀,得到质子导电聚合物a4。其结构式如表2所示。
[0105]
将上述质子导电聚合物a4(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0106]
实施例5
[0107]
odads5atz(8.79g,16.7mmol)在室温、氮气气氛下溶于间甲酚(110ml)、三乙胺(4.21g,41.6mmol)混和溶剂中,向反应瓶中加入ntda(4.60g,17.2mmol)及苯甲酸(3.05g,25mmol),将反应体系加热至80℃,并在此温度下反应4小时,然后再升至180℃,并反应16小时得到预聚物溶液1;将oda(3.34g,16.7mmol)在室温、氮气气氛下溶于间甲酚(60ml)、三乙胺(4.21g,41.6mmol)混和溶剂中,向反应瓶中加入ntda(4.33g,16.2mmol)及苯甲酸(3.05g,25mmol),将反应体系加热至80℃,并在此温度下反应4小时,然后再升至180℃,并反应16小时,得到预聚物溶液2;将预聚物溶液2加入预聚物溶液1中,继续在180℃反应10小时,反应液用丙酮沉淀,得到质子导电聚合物a5。其结构式如表2所示。
[0108]
将上述质子导电聚合物a5(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0109]
实施例6
[0110]
bfsds3stz(4.82g,8.3mmol)、bfs(2.11g,8.3mmol)、ddbp(3.57g,16.7mmol)一起加入50ml三口烧瓶,并加入无水碳酸钾(5.06g,36.6mmol)作为催化剂,nmp(60ml)作为溶剂,甲苯10ml脱水剂加入机械搅拌,接油水分离器和冷凝管,氮气置换三次后,保持氮气流通0.1l/min,在160℃脱水4小时后,升温至180℃反应2小时后结束反应。将反应溶液用500ml异丙醇沉淀,并用100ml异丙醇洗净2次后,80℃真空干燥16小时,得到质子导电聚合物a6。其结构式如表2所示。
[0111]
将上述质子导电聚合物a6(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板
玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0112]
实施例7
[0113]
bfsds3stz(4.82g,8.3mmol)、ddbp(1.82g,8.5mmol)一起加入50ml三口烧瓶,并加入无水碳酸钾(2.58g,18.7mmol)作为催化剂,nmp(40ml)作为溶剂,甲苯5ml脱水剂加入机械搅拌,接油水分离器和冷凝管,氮气置换三次后,保持氮气流通0.1l/min,在160℃脱水4小时后,升温至180℃反应2小时后结束反应,获得预聚物溶液1。bfs(2.11g,8.3mmol)、ddbp(1.75g,8.2mmol)一起加入50ml三口烧瓶,并加入无水碳酸钾(2.58g,18.7mmol)作为催化剂,nmp(20ml)作为溶剂,甲苯5ml脱水剂加入机械搅拌,接油水分离器和冷凝管,氮气置换三次后,保持氮气流通0.1l/min,在160℃脱水4小时后,升温至180℃反应2小时后结束反应得到预聚物溶液2。将预聚物溶液1和预聚物溶液2混合后,继续在180℃反应2小时后,溶液用500ml异丙醇沉淀,并用100ml异丙醇洗净2次后,80℃真空干燥16小时,得到质子导电聚合物a7。其结构式如表2所示。
[0114]
将上述质子导电聚合物a7(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0115]
实施例8
[0116]
dfbpts3stz(7.22g,8.3mmol)、ddbp(1.82g,8.5mmol)一起加入50ml三口烧瓶,并加入无水碳酸钾(2.58g,18.7mmol)作为催化剂,nmp(60ml)作为溶剂,甲苯10ml脱水剂加入机械搅拌,接油水分离器和冷凝管,氮气置换三次后,保持氮气流通0.1l/min,在160℃脱水4小时后,升温至180℃反应4小时后结束反应,获得预聚物溶液1。dfbp(1.82g,8.3mmol)、ddbp(1.75g,8.2mmol)一起加入50ml三口烧瓶,并加入无水碳酸钾(2.58g,18.7mmol)作为催化剂,nmp(20ml)作为溶剂,甲苯5ml脱水剂加入机械搅拌,接油水分离器和冷凝管,氮气置换三次后,保持氮气流通0.1l/min,在160℃脱水4小时后,升温至180℃反应2小时后结束反应得到预聚物溶液2。将预聚物溶液1和预聚物溶液2混合后,继续在180℃反应2小时后,溶液用500ml异丙醇沉淀,并用100ml异丙醇洗净2次后,80℃真空干燥16小时,得到质子导电聚合物a8。其结构式如表2所示。
[0117]
将上述质子导电聚合物a8(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0118]
比较例1
[0119]
bfsds(3.45g,8.3mmol)、ddbp(1.82g,8.5mmol)一起加入50ml三口烧瓶,并加入无水碳酸钾(2.58g,18.7mmol)作为催化剂,nmp(40ml)作为溶剂,甲苯5ml脱水剂加入机械搅拌,接油水分离器和冷凝管,氮气置换三次后,保持氮气流通0.1l/min,在160℃脱水4小时后,升温至180℃反应2小时后结束反应,获得预聚物溶液1。bfs(2.11g,8.3mmol)、ddbp(1.75g,8.2mmol)一起加入50ml三口烧瓶,并加入无水碳酸钾(2.58g,18.7mmol)作为催化剂,nmp(20ml)作为溶剂,甲苯5ml脱水剂加入机械搅拌,接油水分离器和冷凝管,氮气置换三次后,保持氮气流通0.1l/min,在160℃脱水4小时后,升温至180℃反应2小时后结束反应得到预聚物溶液2。将预聚物溶液1和预聚物溶液2混合后,继续在180℃反应2小时后,溶液用500ml异丙醇沉淀,并用100ml异丙醇洗净2次后,80℃真空干燥16小时,得到质子导电聚合物b1.其结构式如表2所示。
[0120]
将上述质子导电聚合物b1(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0121]
比较例2
[0122]
bfpstzt(4.32g,8.3mmol)、ddbp(1.82g,8.5mmol)一起加入50ml三口烧瓶,并加入无水碳酸钾(2.58g,18.7mmol)作为催化剂,nmp(40ml)作为溶剂,甲苯5ml脱水剂加入机械搅拌,接油水分离器和冷凝管,氮气置换三次后,保持氮气流通0.1l/min,在160℃脱水4小时后,升温至180℃反应2小时后结束反应,获得预聚物溶液1。bfs(2.11g,8.3mmol)、ddbp(1.75g,8.2mmol)一起加入50ml三口烧瓶,并加入无水碳酸钾(2.58g,18.7mmol)作为催化剂,nmp(20ml)作为溶剂,甲苯5ml脱水剂加入机械搅拌,接油水分离器和冷凝管,氮气置换三次后,保持氮气流通0.1l/min,在160℃脱水4小时后,升温至180℃反应2小时后结束反应得到预聚物溶液2。将预聚物溶液1和预聚物溶液2混合后,继续在180℃反应2小时后,溶液用500ml异丙醇沉淀,并用100ml异丙醇洗净2次后,80℃真空干燥16小时,得到质子导电聚合物b2。其结构式如表2所示。
[0123]
将上述质子导电聚合物b2(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0124]
比较例3
[0125]
odads(6.00g,16.7mmol)、baoz(4.20g,16.7mmol)在室温、氮气气氛下溶于间甲酚(150ml)、三乙胺(8.43g,83.3mmol)混和溶剂中,向反应瓶中加入ntda(9.20g,34.3mmol)及苯甲酸(6.10g,50mmol),将反应体系加热至80℃,并在此温度下反应4小时,然后再升至180℃,并反应16小时,反应液用丙酮沉淀,得到质子导电聚合物b3。其结构式如表2所示。
[0126]
将上述质子导电聚合物b3(5g)溶解于25ml nmp中,将此溶液浇注在12
×
12cm平板玻璃上,于50℃真空干燥16小时,然后在160℃真空干燥3小时后,将膜用ipa浸泡洗去残留的有机溶剂后,用10%的稀硫酸浸泡48小时后,再用去离子水洗至中性,真空烘干得到聚合物电解质膜,膜厚10μm,聚合物电解质膜的相对湿度90rh%时随温度变化的质子导电率见图1,arrhenius曲线见图2,热水溶胀度、质子导电活化能和120℃随湿度变化的质子导电率见表3。
[0127]
表2实施例和比较例的质子导电性聚合物
[0128]
[0129][0130]
表3实施例和比较例的质子导电性聚合物的性能
[0131][0132]
含有磺酰胺基和与磺酰胺基共价键连接的含氮杂环化合物的实施例1~实施例8的热水溶胀度都在5%以下,相比于只含有磺酸基,磺酸基没有共价键连接含氮杂环化合物的质子导电聚合物b1(比较例1),热水溶胀度显著降低了。这是由于实施例1~实施例8的质子导电聚合物结构中的磺酰基团被共价键连接了胺基和含氮杂环化合物后,吸水溶胀后的尺寸稳定性增加了。相对的含有磺酸基和聚合物骨架中含有含氮杂环化合物的质子导电聚合物b3(比较例3)的热水溶胀度为7.8%,仍然高于实施例1~实施例8的热水溶胀度。
[0133]
含有磺酰胺基及共价键连接的含氮杂环化合物的质子导电聚合物a7(实施例7),在120℃,相对湿度90rh%时的质子导电率为0.041s/cm,根据arrhenius曲线计算得到的质子导电活化能为12.9kj/mol,当相对湿度降到30rh%时,其质子导电率为0.034s/cm。具有
与a7相同聚合物骨架但只含有磺酸基,没有共价键连接含氮杂环化合物的质子导电聚合物b1(比较例1),在120℃,相对湿度90rh%时的质子导电率为0.012s/cm,根据arrhenius曲线计算得到的质子导电活化能为32.1kj/mol,当相对湿度降到30rh%时,其质子导电率为0.003s/cm。与a7相同聚合物骨架但是含有磺酰基和共价键连接的含氮杂环化合物的质子导电聚合物b2(比较例2),在120℃,相对湿度90rh%时的质子导电率为0.021s/cm,根据arrhenius曲线计算得到的质子导电活化能为25.8kj/mol,当相对湿度降到30rh%时,其质子导电率为0.009s/cm。实施例7相对于比较例1和比较例2具有更高的质子导电率和更低的质子导电活化能;且当相对湿度降低时,质子导电率的下降幅度更低。这是因为含有磺酰胺基及共价键连接的含氮杂环化合物的结构,磺酰胺基具有按照vehicle机理传导质子的能力,磺酰胺基的吸电子作用以及其仲胺键共价键连接到含氮杂环化合物上又能进一步发挥出了含氮杂环化合物按照grotthuss机理进行质子传导的潜力。
[0134]
含有磺酰胺基及共价键连接的含氮杂环化合物的质子导电聚合物a1(实施例1),在120℃,相对湿度90rh%时的质子导电率为0.030s/cm,根据arrhenius曲线计算得到的质子导电活化能为20.0kj/mol,当相对湿度降到30rh%时,其质子导电率为0.020s/cm。含有磺酸基,但是含氮化合物不与磺酸相连接而是包含在聚合物骨架中的质子导电聚合物b3(比较例3),在120℃,相对湿度90rh%时的质子导电率为0.019s/cm,根据arrhenius曲线计算得到的质子导电活化能为28.2kj/mol,当相对湿度降到30rh%时,其质子导电率为0.008s/cm。实施例1相对于比较例3具有相近的聚酰亚胺骨架结构,但具有更高的质子导电率和更低的质子导电活化能;且当相对湿度降低时,质子导电率的下降幅度更低。
[0135]
与含有吡唑基的含氮杂环化合物的质子导电聚合物a1(实施例1),具有相同聚合物骨架结构,分别含有1,2,4-三氮唑、1,2,3,4-四氮唑的质子导电聚合物a2和a4(实施例2,实施例4)具有更高的质子导电率和更低的质子导电活化能,因此优选。质子导电聚合物a2(实施例2)含有的1,2,4-三氮唑且杂环中同时含有n-h键和未与h结合的n原子,相比于具有相同聚合物骨架也含有的1,2,4-三氮唑但杂环中只含有未于h结合的n原子的质子导电聚合物a3(实施例3),具有更高的质子导电率和更低的质子导电活化能,因此优选。质子导电聚合物a5和a7(实施例5和实施例7)为嵌段聚合物结构,相比于具有相同组成单元但为无规聚合物的a4和a6(实施例4和实施例6)具有更高的质子导电率和更低的质子导电活化能和更低的热水溶胀度,因此优选。
[0136]
在组成结构中含有更高含磺酰胺基和含氮杂环化合物数量的质子导电聚合物a8(实施例8)具有最高的质子导电率和最低的质子导电活化能。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。