制备芜地溴铵的方法
1.本技术是2017年11月10日提交的发明名称为“制备芜地溴铵的方法”的中国专利申请201780081759.9的分案申请。
2.引言
3.本发明涉及制备4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(一种已知名为芜地溴铵(umeclidinium bromide)的化合物)的新方法。
[0004]
芜地溴铵是有效的抗胆碱药并且已用于治疗呼吸系统疾病诸如哮喘或慢性阻塞性肺病(copd)。它用于制备药物组合物,该药物组合物作为供口腔吸入的干粉以每日一次微克剂量来给药。本发明开发了新的组合物、组合、给药形式(例如定量吸入器)以及使用芜地溴铵的剂量。
[0005]
发明背景
[0006]
以下描述的分子结构(i)的化合物芜地溴铵是用于治疗患有慢性阻塞性肺病(copd)(包括慢性支气管炎和肺气肿)的患者的气道阻塞的长效毒蕈碱拮抗剂。
[0007][0008]
wo2005/104745中已经要求保护了芜地溴铵的合成,其包括如下四个步骤:
[0009]
[0010]
作为关键中间体的式(ii)的1-(2-氯乙基)哌啶-4-羧酸乙酯是通过在碳酸钾的存在下使1-溴-2-氯乙烷和哌啶-4-羧酸乙酯(ethyl isonipecotate)在丙酮中反应来合成。然而,由于二聚体副产物1,1'-(乙烷-1,2-二基)双(哌啶-4-羧酸)二乙酯(v)(其必须通过色谱技术从主要化合物分离)的形成,式(ii)的化合物以非常低的收率(39%)制备。
[0011][0012]
为了克服二聚作用问题和随之而来的低收率,wo2014/027045要求保护了一种以较好收率(80%)制备式(ii)化合物的替代两步法,如下所示:
[0013][0014]
毫无疑问,这样的合成替代方案可以得出更好的收率,但是需要两个反应步骤而非wo2005/104745中描述的单个反应步骤,因而不是工业应用的最佳方案。另外,wo2014/027045公开了在第一步中使用高温并且在第二步中使用具有高腐蚀性和高毒性的试剂,即,产生不环保的sox副产物的亚硫酰氯。当与wo2005/104745中描述的温和条件相比时,具有三个主要缺点。
[0015]
替代地,wo2016/071792要求保护制备式(ii)的化合物的一步方法,其包括将哌啶-4-羧酸乙酯与卤代乙醛在甲醇:乙酸以及还原剂的混合物中进行反应,如下所示:
[0016]
[0017]
尽管与wo2005/104745和wo2014/027045中描述的那些方法相比得到更好的收率(90%),但该合成需要使用甲醇-水酸性溶液,在与还原剂反应之前,该溶液可在一定程度上降解酯基团。
[0018]
wo2011/029896描述了一种通过使用不同中间体来制备芜地溴铵的替代方法,如下所示:
[0019][0020]
其中p是保护基;r选自烷基、烯基、炔基、环烷基、环烯基、杂环、芳基和杂芳基;并且x和y是离去基团,条件是x和y不同。
[0021]
然而,该方法比wo2005/104745中公开的方法更复杂,因为它包括更长的合成路线并且包括额外的保护-脱保护步骤。
[0022]
芜地溴铵的非溶剂化结晶形式已经公开为活性药物成分的多晶型物(wo2014/027045、us9273001 b2),表明该化合物可以产生具有不同物理性质的多种固体。制备单晶形式的纯芜地溴铵对于本行业来说是一个挑战,因为芜地溴铵对于形成溶剂合物是高度敏感的。芜地溴铵溶剂合物包括甲醇溶剂合物(cz27764(赛诺菲)),并且已经公开了乙醇、2-丙醇、2-甲基丙-1-醇、氯苯和对二甲苯溶剂合物(wo2014/027045、us9273001b2)。在最终工艺步骤中将1-丙醇用作溶剂以最小化溶剂合物的形成(us 9273001 b2),避免了先前需要的化合物在乙酸乙酯、甲醇和水中的再悬浮(实施例84,方法b,wo2005/104745)。
[0023]
为了满足芜地溴铵市场需求,需要开发更有效的方法。即,提供优于先前公开在wo2016/071792、wo2005/104745、wo2014/027045和wo2011/029896中的那些方法的优点的方法。还需要提供制备具有一致的结晶度和化学纯度水平的单一纯结晶形式的芜地溴铵的方法。
技术实现要素:
[0024]
根据本发明的一个方面,提供了一种制备芜地溴铵的方法,该方法包括:
[0025]
a)在有机碱的存在下,将哌啶-4-羧酸乙酯与1-溴-2-氯乙烷在溶剂中反应,以形成1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)或其盐;
[0026]
b)将1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)或其盐与二异丙基氨基锂在溶剂中反
应,以形成1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii);
[0027]
c)将1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii)与苯基锂在溶剂中反应,以形成1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv);以及
[0028]
d)将1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv)与((2-溴乙氧基)甲基)苯在溶剂中反应,以形成4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i),即,芜地溴铵。
[0029]
根据本发明的另一方面,提供了一种方法,该方法包括:
[0030]
a)在有机碱的存在下,将哌啶-4-羧酸乙酯与1-溴-2-氯乙烷在溶剂中反应,以形成1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)或其盐。
[0031]
根据本发明的又一方面,提供了一种方法,该方法包括:
[0032]
d)将1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇与((2-溴乙氧基)甲基)苯在溶剂中反应,以形成4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i),即,芜地溴铵,其中所述溶剂选自环醚(诸如四氢呋喃)、芳族溶剂(诸如甲苯)、酮(诸如丙酮)、和质子溶剂(诸如水)、或其组合,任选地,其中所述溶剂是水。
[0033]
本发明的其它方面涉及通过本发明的方法可获得的1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)和芜地溴铵,以及包含所述芜地溴铵的药物组合物。
[0034]
令人惊奇地,已经发现,本发明的步骤a)以比wo2005/104745中公开的方法更高的收率(66%)提供关键中间体1-(2-氯乙基)哌啶-4-羧酸乙酯(ii),而不需要增加工艺步骤(诸如保护-脱保护步骤)的数量,不需要使用高温并且不需要使用不期望的试剂(诸如腐蚀性试剂、毒性试剂或甲醇/水酸性体系)。本发明的步骤a)控制不期望的副产物诸如1,1'-(乙烷-1,2-二基)双(哌啶-4-羧酸)二乙酯(v)的形成。在本发明的步骤a)期间获得的1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)可以纯化或者无需纯化(例如,通过色谱纯化)而直接用于接下来工艺步骤。本发明的方法实现了芜地溴铵的叠缩(telescoped)(或一锅法)合成,由此使原料经历连续的化学反应。这样的合成是非常需要的,因为它通过避免中间体的分离和纯化而提高了化学反应效率,从而节省了时间和资源,同时提高了化学收率。
[0035]
本发明的步骤a)的一个优点是使用从该工艺步骤获得的1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)中间体使得能够制备具有一致的结晶度和化学纯度水平的单一纯结晶形式的芜地溴铵。
[0036]
另外,已经发现,本发明的步骤d)提供了具有一致的结晶度和化学纯度水平的单一纯结晶形式的产品。因此,本发明的方法的又一个优点是在本发明的步骤d)中获得的芜地溴铵是具有一致的结晶度和化学纯度水平的单一纯结晶形式。
[0037]
因此,本发明公开了制备提供具有一致的结晶度和化学纯度水平的单一纯结晶形式的芜地溴铵的方法。
[0038]
最后,本发明的方法实现了具有适于吸入的粒度的芜地溴铵的生产。
附图说明
[0039]
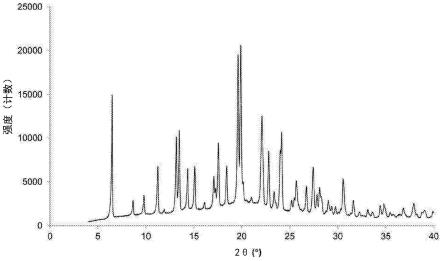
图1:从实施例18获得的芜地溴铵的xrpd衍射图。
[0040]
图2:从实施例18获得的芜地溴铵的dsc热谱图。
[0041]
图3:从实施例18获得的芜地溴铵的tga热谱图。
[0042]
图4:从实施例18获得的芜地溴铵的hplc。
[0043]
图5:在实施例19的三个步骤之后获得的并且在最终步骤中用作制备芜地溴铵的原料的1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇的hplc。
[0044]
图6:来自实施例20的从水中重结晶的芜地溴铵的hplc
[0045]
图7:微粉化之前的芜地溴铵的xrpd。
[0046]
图8:来自实施例21的流体能量喷射磨微粉化后的芜地溴铵的xrpd。
[0047]
图9:来自实施例22的高压匀浆微粉化后的芜地溴铵的xrpd。
[0048]
发明详述
[0049]
本发明提供了另一种制备芜地溴铵和用于制备芜地溴铵的关键中间体之一(即,1-(2-氯乙基)哌啶-4-羧酸乙酯(ii))的方法。
[0050]
本发明可提供一种方法,其包括以下步骤:
[0051]
a)在有机碱的存在下,将哌啶-4-羧酸乙酯与1-溴-2-氯乙烷在溶剂中反应,以形成1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)或其盐;
[0052]
b)将1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)或其盐与二异丙基氨基锂在溶剂中反应,以形成1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii);
[0053]
c)将1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii)与苯基锂在溶剂中反应,以形成1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv);
[0054]
d)将1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv)与((2-溴乙氧基)甲基)苯在溶剂中反应,以形成单一纯结晶形式的4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i),即,芜地溴铵,以及任选地;
[0055]
e)使芜地溴铵重结晶,以始终如一地获得高纯度产物,以及任选地;
[0056]
f)将芜地溴铵微粉化以获得具有适合于吸入的粒度同时保持其结晶形式的产物。
[0057]
步骤a)至e)可以在不存在步骤f)的情况下组合。步骤a)至d)和f)可以在不存在步骤e)的情况下组合。
[0058]
步骤d)和e)可以在不存在其他工艺步骤的情况下组合。步骤d)和f)可以在不存在其他工艺步骤的情况下组合。步骤d)、e)和f)可以在不存在其他工艺步骤的情况下组合。
[0059]
本发明的步骤a)可按如下进行:
[0060]
a)在约20℃至约56℃的温度下,在有机碱的存在下,将哌啶-4-羧酸乙酯与1-溴-2-氯乙烷在溶剂中反应,以形成1-(2-氯乙基)哌啶-4-羧酸乙酯(ii);以及优选地在此后(i)进行溶剂更换(exchange),从反应混合物中除去形成的任何盐(优选地通过水萃取进行),并在过滤后浓缩所得溶液以从溶液中分离存在的1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)从而除去二聚物;或此后(ii)从反应混合物中除去形成的任何盐(优选地通过水萃取,用无机酸或有机酸(优选盐酸、乙酸、琥珀酸或草酸)酸化所得溶液来进行),并以盐的形式分离产物1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)(优选地通过过滤和干燥进行)。
[0061]
步骤a)中使用的溶剂可以选自酮诸如丙酮。
[0062]
步骤a)中使用的有机碱可以选自诸如胺类的有机碱,如三乙胺、吡啶、n,n-二异丙基乙胺、4-(二甲基氨基)吡啶、1,8-二氮杂双环[5.4.0]十一碳-7-烯。优选地,所述有机碱是三乙胺。在完成步骤a)的反应后,可进行溶剂更换,可通过水萃取除去三乙胺盐,并可将所得溶液浓缩以分离1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)。通过使用三乙胺作为有机碱,可
以高达66%的收率获得1-(2-氯乙基)哌啶-4-羧酸乙酯(ii),其中1,1'-(乙烷-1,2-二基)双(哌啶-4-羧酸)二乙酯(v)的残留含量低于14%。相比之下,通过进行wo2005/104745中公开的发明人的操作,副产物1,1'-(乙烷-1,2-二基)双(哌啶-4-羧酸)二乙酯(v)的形成达到22%。
[0063]
步骤a)可在约20℃至约56℃,优选约20℃至约30℃的温度下进行,更优选地,该反应在约20℃至约25℃的温度下进行。在高于30℃的温度下,获得更大量的副产物1,1'-(乙烷-1,2-二基)双(哌啶-4-羧酸)二乙酯(v),以较低收率(34%)获得1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)。步骤a)可以进行约14h至约24h的时间段。
[0064]
在通过水萃取除去任何盐(例如三乙胺盐)之后,可以用无机酸或有机酸(优选使用盐酸、乙酸、琥珀酸或草酸或其溶液)酸化所得溶液,并且可以以盐的形式分离产物1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)(优选通过过滤和干燥进行)。
[0065]
如上所述,步骤a)可包括更换反应溶剂。更换溶剂可包括一种或多种烷烃,例如正庚烷或庚烷的混合物。
[0066]
如上文还公开的,步骤a)可包括通过过滤除去二聚体。可以在过滤之前将反应混合物冷却,任选地冷却至约-20℃,并且在该温度下保持约12h至约24h,任选地约16h。
[0067]
组合起来说,步骤a)可以包括:
[0068]
i)更换反应溶剂;
[0069]
ii)水萃取;和
[0070]
iii)通过过滤除去二聚体。
[0071]
本发明的步骤b)可按如下进行:
[0072]
b)优选在约-50℃至约25℃的温度下,将1-(2-氯乙基)哌啶-4-羧酸乙酯(ii)或其盐与二异丙基氨基锂在溶剂中反应,以形成1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii)或其盐;并且优选地在此后从反应混合物中除去形成的任何盐(优选地通过碱性水萃取进行),并且进行溶剂蒸馏和溶剂更换。
[0073]
步骤b)中使用的溶剂可以选自环醚诸如四氢呋喃(thf)。
[0074]
本发明的步骤c)可按如下进行:
[0075]
c)优选在约-30℃至约25℃的温度下,将1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii)或其盐与苯基锂在溶剂中反应,以形成1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇或其盐;优选地用水处理含有1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv)或其盐的反应混合物,并浓缩所得溶液,并且优选地在此后加入合适的反溶剂以实现沉淀,并且分离如此形成的产物(优选地通过过滤和干燥进行),经hplc测定其纯度≥98.0%。
[0076]
步骤c)中使用的溶剂可以选自环醚诸如thf。
[0077]
本发明的步骤d)可按如下进行:
[0078]
d)在约40℃至约溶剂回流温度的温度下,优选在约60℃至约溶剂回流温度的温度下,将1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv)与(2-溴乙氧基)甲基)苯在溶剂中反应,以形成4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i),即,芜地溴铵,并且优选地将反应混合物冷却至约-10℃至约5℃的温度,优选地在约-10℃至约5℃的温度下将悬浮液搅拌约2h。此后,可以分离所得产物(优选通过过滤进行)并在约35℃至约55℃的温度下干燥(优选在真空下),该产物经hplc测定纯度≥
98.0%,且为单晶形式。
[0079]
步骤d)中使用的溶剂可选自环醚(诸如thf)、芳族溶剂(诸如甲苯)、酮(诸如丙酮)、和质子溶剂(诸如水)。步骤d)可以在约40℃至约111℃,优选地任选地约60℃至约100℃的温度下进行。优选地,在约60℃至约100℃的温度下,在水中进行该反应。步骤d)可以进行约18h时至约24h的时间段。当反应完成时,将反应冷却,使得能够以高达的84%的收率获得芜地溴铵。通过遵循所描述的操作获得的产物的纯度通常经hplc测定为≥98.0%,且为单晶形式。分离的芜地溴铵的结晶形式是芜地溴铵的非溶剂化形式。
[0080]
本发明的步骤e)可按如下进行:
[0081]
e)在约40℃至约溶剂回流温度的温度下,优选在约60℃至约80℃的温度下,将芜地溴铵在溶剂中重结晶,以形成4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i),即,芜地溴铵,并且优选将反应混合物冷却至约-10℃至约5℃的温度,优选地在约-10℃至约5℃的温度下将悬浮液搅拌约2h。此后,分离所得产物(优选通过过滤进行)并在约35℃至约55℃的温度下干燥(优选在真空下),以提供经hplc测定纯度≥99.0%且为单晶形式的产物。
[0082]
可以对根据本发明的步骤d)获得的芜地溴铵进行重结晶。重结晶溶剂可以选自醇(如1-丙醇)、质子溶剂(诸如水)、或这两类溶剂的混合物。优选地,在水中进行重结晶,任选地通过在约40℃至约溶剂回流温度的温度下,优选在约60℃至约80℃的温度下将材料悬浮在水中来进行重结晶。可将所得溶液冷却至约-10℃至约5℃的温度,并且可将所得悬浮液在约-10℃至约5℃的温度下搅拌约2小时。优选地,分离芜地溴铵(任选通过过滤进行),用水洗涤(任选地),然后干燥。可以在约35℃至约55℃的温度下真空干燥芜地溴铵。干燥的产物通常具有经hplc测定≥99.0%的纯度,并表现出单晶形式。
[0083]
图1-9中示出了根据本发明获得的产物的x射线粉末衍射(xrpd)衍射图、差示扫描量热法(dsc)热谱图、热重分析(tga)热谱图和hplc色谱图。
[0084]
优选将从本发明中获得的芜地溴铵微粉化以获得具有适于吸入的粒度的材料。因此,本发明还提供了一种用于调整粒度并同时保持芜地溴铵的晶型的微粉化方法。
[0085]
提供以下实施例以说明本发明的方法,所述实施例并不旨在解释为对本发明的限制;在不脱离本发明的精神和范围的情况下,可以采取略微的变化。
[0086]
实施例1
[0087]
1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)的制备
[0088]
将三乙胺(1.09ml,7.79mmol)添加到哌啶-4-羧酸乙酯(0.80ml,5.19mmol)的丙酮(7.20ml)溶液中,随后加入1-溴-2-氯乙烷(0.86ml,10.38mmol)。将反应混合物在25℃下搅拌24h,然后在真空下浓缩。将所得残余物用水(3.0ml)处理,并用乙酸乙酯(3x3.0ml)萃取。将合并的有机层用mgso4干燥,过滤并在真空下浓缩。通过经硅胶快速层析(梯度1:1正己烷/乙酸乙酯至9:1乙酸乙酯/甲醇)进行粗产物的纯化,得到所需的化合物(无色液体,0.75g,65.6%)和相应二聚体(0.25g,14.0%)。
[0089]
1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii):1h-nmr(300mhz,cdcl3)δ4.11(q,j=5.3hz,2h),3.55(t,j=4.0hz,2h),2.88
–
2.84(m,2h),2.68(t,j=4.0hz,2h),2.24(dt,j=12.0,3.0hz,1h),2.12(td,j=11.3,2.7hz,2h),1.93
–
1.65(m,4h),1.22(t,j=9.0hz,3h);
13
c-nmr(75mhz,cdcl3)δ175.11,60.51,60.21,53.18,41.23,41.09,28.29,14.38。针对c10h18
clno2计算的ms(esi)m/z:219,实测值220[m h]
。
[0090]
1,1'-(乙烷-1,2-二基)双(哌啶-4-羧酸)二乙酯(v):h-nmr(300mhz,cdcl3)δ4.11(q,j=5.25hz,4h),2.99
–
2.76(m,4h),2.47(s,4h),2.30
–
2.20(m,2h),2.07
–
1.99(m,6h),1.90
–
1.84(m,4h),1.79
–
1.66(m,4h),1.23(t,j=6.0hz,6h);
13
c-nmr(75mhz,cdcl3)δ175.29,60.49,56.43,53.68,41.29,28.40,14.40。针对c
18h32
n2o2计算的ms(esi)m/z:340,实测值341[m h]
。
[0091]
实施例2
[0092]
1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)的制备
[0093]
将三乙胺(1.09ml,7.79mmol)添加到哌啶-4-羧酸乙酯(0.80ml,5.19mmol)的丙酮(8.60ml)溶液中,随后加入1-溴-2-氯乙烷(0.86ml,10.38mmol)。将反应混合物在25℃下搅拌17h。然后加入正庚烷(8.6ml),并在真空下除去丙酮。向所得混合物中再加入正庚烷(8.6ml),并在真空下除去更多的丙酮以获得8.6ml的体积。向混合物中加入水(8.6ml),并用正庚烷(2x8.6ml)萃取。将合并的有机层用mgso4干燥,过滤并在真空下浓缩。进一步地,添加正庚烷(2.40ml),并将溶液置于0℃下1h,并冷却至-20℃持续16h。过滤该溶液以除去二聚体(1,1'-(乙烷-1,2-二基)双(哌啶-4-羧酸)二乙酯(v)),然后在真空下浓缩。通过经硅胶快速层析(梯度1:1正己烷/乙酸乙酯至9:1乙酸乙酯/甲醇)进行粗产物的纯化,得到所需的化合物(无色液体,0.63g,55.1%)和相应二聚体(0.10g,5.8%)。
[0094]
实施例3
[0095]
1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)的制备
[0096]
在56℃下,在5h内将哌啶-4-羧酸乙酯(0.40ml,2.60mmol)和三乙胺(0.55ml,3.90mmol)的丙酮(1.3ml)溶液缓慢添加到1-溴-2-氯乙烷(0.43ml,5.19mmol)的丙酮(3.0ml)溶液中。将反应混合物在56℃下搅拌24h,然后在真空下浓缩。将所得残余物用水(1.0ml)处理,并用乙醚(3x3.0ml)萃取。将合并的有机层用mgso4干燥,过滤并在真空下浓缩。通过经硅胶快速层析(6:4正己烷/乙酸乙酯)进行粗产物的纯化,得到所需的化合物(无色液体,0.19g,33.5%)和相应二聚体。
[0097]
实施例4
[0098]
1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)的制备
[0099]
在室温下,在5h内将哌啶-4-羧酸乙酯(0.40ml,2.60mmol)和三乙胺(0.55ml,3.90mmol)的丙酮(1.6ml)溶液缓慢添加到1-溴-2-氯乙烷(0.86ml,10.38mmol)和碘化钾(10%,1.04mmol,0.17mg)的丙酮(7.0ml)溶液中。将反应混合物在25℃下搅拌24h,然后在真空下浓缩。将所得残余物用水(1.0ml)处理,并用乙醚(3x3.0ml)萃取。将合并的有机层用mgso4干燥,过滤并在真空下浓缩。通过经硅胶快速层析(6:4正己烷/乙酸乙酯)进行粗产物的纯化,得到所需的化合物(无色液体,0.29g,50.1%)和相应二聚体。
[0100]
实施例5
[0101]
1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)的制备
[0102]
将n,n-二异丙基乙胺(dipea)(1.36ml,7.79mmol)添加到哌啶-4-羧酸乙酯(0.80ml,5.19mmol)的丙酮(8.60ml)溶液中,随后加入1-溴-2-氯乙烷(0.86ml,10.38mmol)。将反应混合物在25℃下搅拌24h。然后添加水(3.0ml),用hcl(1m)中和ph,并用乙醚(3x10.0ml)萃取水相。将合并的有机层用mgso4干燥,过滤并在真空下浓缩。通过1h-nmr
分析粗产物(0.67g),得到1.00:0.06的1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)与二聚体。
[0103]
实施例6
[0104]
1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)的制备
[0105]
将4-二甲基氨基吡啶(dmap)(0.95g,7.79mmol)添加到哌啶-4-羧酸乙酯(0.80ml,5.19mmol)的丙酮(8.60ml)溶液中,随后加入1-溴-2-氯乙烷(0.86ml,10.38mmol)。将反应混合物在25℃下搅拌24h。然后添加水(3.0ml),用hcl(1m)中和ph,并用乙醚(3x10.0ml)萃取水相。将合并的有机层用mgso4干燥,过滤并在真空下浓缩。通过1h-nmr分析粗产物(0.52g),得到1.00:0.06的1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)与二聚体。
[0106]
实施例7
[0107]
1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)的制备
[0108]
将1,8-二氮杂双环十一碳-7-烯(dbu)(1.16ml,7.79mmol)添加到哌啶-4-羧酸乙酯(0.80ml,5.19mmol)的丙酮(8.60ml)溶液中,随后加入1-溴-2-氯乙烷(0.86ml,10.38mmol)。将反应混合物在25℃下搅拌24h。然后添加水(3.0ml),用hcl(1m)中和ph,并用乙醚(3x10.0ml)萃取水相。将合并的有机层用mgso4干燥,过滤并在真空下浓缩。通过1h-nmr分析粗产物(0.88g),未检测到二聚体。
[0109]
实施例8
[0110]
1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)的制备
[0111]
将吡啶(0.63ml,7.79mmol)添加到哌啶-4-羧酸乙酯(0.80ml,5.19mmol)的丙酮(8.60ml)溶液中,随后加入1-溴-2-氯乙烷(0.86ml,10.38mmol)。将反应混合物在25℃下搅拌24h。然后添加正庚烷(8.6ml),并在真空下除去丙酮。向所得混合物中再加入正庚烷(8.6ml),并在真空下除去更多的丙酮以获得8.6ml的体积。向混合物中添加水(8.6ml),并用正庚烷(2x8.6ml)萃取。将合并的有机层用mgso4干燥,过滤并在真空下浓缩。通过1h-nmr分析粗产物(0.38g),得到1.00:0.10比率的1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)与二聚体。
[0112]
实施例9
[0113]
1-(2-氯乙基)-4-哌啶-4-羧酸乙酯盐酸盐的制备
[0114]
在室温下向1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)(0.15ml)的乙酸乙酯(2ml)溶液中逐滴加入氯化氢(1.25m)的乙醇(0.72ml)溶液。在真空下除去溶剂,得到结晶白色固体。
[0115]
实施例10
[0116]
1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii)的制备
[0117]
将1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)(5.0g,22.76mmol)的四氢呋喃(thf,147.0ml)溶液在氮气下冷却至-50℃。在-50℃下,将lda(在庚烷/thf/乙苯中2.0m,17.0ml,34.0mmol)在25分钟内添加到溶液中。使反应混合物在16h内升温至室温。用饱和k2co3水溶液(122.0ml)终止反应,并用乙醚(3x120.0ml)萃取。将合并的有机层用mgso4干燥,过滤并在真空下浓缩。用二氯甲烷将得到的橙色液体共蒸发三次以除去过量的乙苯,得到橙色油状物(4.15g,99.4%)。
[0118]
1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii):1h-nmr(300mhz,cdcl3)δ4.10(t,j=5.23hz,2h),2.90
–
2.85(m,6h),1.71
–
1.66(m,6h),1.22(t,j=4.0hz,3h)。
[0119]
实施例11
[0120]
1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv)的制备
[0121]
将苯基锂的溶液(在70环己烷/30乙醚中1.9m,22.30ml,42.40mmol)在氮气下冷却至-30℃。在-30℃下,在25分钟内将1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii,2.0g,10.90mmol)的thf(27.0ml)溶液缓慢添加到反应混合物中。使反应混合物在16h内升温至室温。用水(10.0ml)终止反应,然后在真空下蒸发至干燥。添加水(40.0ml)和乙酸乙酯(40.0ml),使白色固体出现。在真空下滤出固体,得到白色粉末(2.46g,76.8%)。
[0122]
1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv):1h-nmr(300mhz,cdcl3)δ7.54
–
7.51(m,3h),7.33
–
7.20(m,6h),2.85
–
2.80(m,6h),1.78
–
1.72(m,6h)。针对c
20h23
no计算的ms(esi)m/z:293,实测值294[m h]
。
[0123]
实施例12
[0124]
4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i)的制备
[0125]
向1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv,0.20g,0.69mmol)的thf(30.0ml)溶液中添加((2-溴乙氧基)甲基)苯(0.16ml,1.03mmol)。将溶液在60℃下搅拌24h。然后将溶液冷却至25℃并在真空下浓缩,形成白色固体。过滤该产物并用乙酸乙酯(5x20.0ml)和正己烷(5x20.0ml)在真空下洗涤。然后在真空下干燥白色固体(0.30g,82.2%)。
[0126]
4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i):1h-nmr(300mhz,dmso-d6)δ7.54(d,j=6.0hz,4h),7.35
–
7.20(m,11h),5.97(s,1h),4.49(s,2h),3.81(b,2h),3.49
–
3.46(m,6h),3.31(s,2h),1.99(bt,j=6.0hz,6h)。针对c
29h34
no2计算的ms(esi)m/z:428,实测值428[m h]
。
[0127]
实施例13
[0128]
4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i)的制备
[0129]
向1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv,0.20g,0.69mmol)在丙酮(30.0ml)中的悬浮液中加入((2-溴乙氧基)甲基)苯(0.16ml,1.03mmol)。在60℃下将反应混合物搅拌24h。然后将反应混合物冷却至25℃并在真空下浓缩,形成白色固体。过滤该产物并用乙酸乙酯(5x20.0ml)和正己烷(5x20.0ml)在真空下洗涤。然后在真空下干燥白色固体(0.27g,75.7%)。
[0130]
实施例14
[0131]
4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i)的制备
[0132]
向1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv,0.20g,0.69mmol)在甲苯(30.0ml)中的悬浮液中加入((2-溴乙氧基)甲基)苯(0.16ml,1.03mmol)。在60℃下将反应混合物搅拌24h。然后将溶液冷却至25℃并在真空下浓缩,形成白色固体。过滤该产物并用乙酸乙酯(5x20.0ml)和正己烷(5x20.0ml)在真空下洗涤。然后在真空下干燥白色固体(0.28g,79.6%)。
[0133]
实施例15
nmr分析该第一粗产物(4.02g),得到1.00:0.11比率的1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)与二聚体。进一步地,将正庚烷(11.50ml)添加到粗产物中,并将溶液置于0℃下1h,冷却至-20℃持续16h。过滤固体以除去二聚体,然后在真空下浓缩1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)的溶液。通过1h-nmr分析该第二粗产物(3.57g),得到1.00:0.09比率的1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)与二聚体。
[0148]
将第二粗产物(1-(2-氯乙基)-4-哌啶-4-羧酸乙酯(ii)3.57g)的thf(89.6ml)溶液在氮气下冷却至-50℃。在-50℃下,将lda(在己烷/thf中1.0m,20.72ml,20.72mmol)在25分钟内添加到溶液中。使反应混合物在16h内升温至室温。用饱和k2co3水溶液(74.4ml)终止反应,并用乙酸乙酯(3
×
74.4ml)萃取。将合并的有机层用mgso4干燥,过滤并在真空下浓缩,得到作为橙色油状物的粗品1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii)(3.02g)。
[0149]
将苯基锂(在70环己烷/30乙醚中1.9m,33.7ml,64.1mmol)的溶液在氮气下冷却至-30℃。在-30℃下,在25分钟内将粗品1-氮杂二环[2.2.2]辛烷-4-羧酸乙酯(iii,3.02g)的thf(36.7ml)溶液缓慢添加到反应混合物中。使反应混合物在16h内升温至室温。用水(15ml)终止反应,然后在真空下蒸发至干燥(结果:黄色固体)。添加水(60.2ml)和乙酸乙酯(60.2ml),使白色固体出现。在真空下滤出该固体,得到作为白色粉末的粗品1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv)(1.76g,三步收率:23.0%)。
[0150]
向粗品1-氮杂二环[2.2.2]辛-4-基(二苯基)甲醇(iv,1.76g,5.83mmol)在水(258.0ml)中的悬浮液中加入((2-溴乙氧基)甲基)苯(1.40ml,9.01mmol)。在回流下将反应混合物搅拌24h。然后将反应混合物缓慢冷却至2℃-4℃的温度,并在2℃-4℃的温度下搅拌2h。在真空下过滤产物,通过用庚烷(20.0ml)洗涤化合物除去过量的溴化物。然后将白色固体在真空下干燥(2.55g,最终步骤收率84.0%)。
[0151]
实施例20
[0152]
4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i)的重结晶
[0153]
将4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i,0.20g)在水(2.25ml)中的悬浮液加热至80℃。将溶液搅拌1h,然后缓慢冷却至2℃-4℃的温度,并在2℃-4℃的温度下搅拌2h。过滤固体并在真空下干燥(0.185g,92.5%)。
[0154]
实施例21
[0155]
4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i)的微粉化
[0156]
将4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i,3.0g)以10g/h的速率投料到流体能量喷射磨中,该流体能量喷射磨在针对文丘里流量计为4巴的压力下且针对环为3巴的压力下用n2进行操作。
[0157]
分离的产物展示出与具有以下粒度分布的原料相同的xrpd:dv10=0.664μm;dv50=3.071μm;dv90=7.013μm;跨度=2.07,如图8所示。
[0158]
实施例22
[0159]
4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i)的微粉化
[0160]
将4-[羟基(二苯基)甲基]-1-[2-(苯基甲基)氧基]乙基]-1-氮鎓杂二环[2.2.2]辛烷溴化物(i,15.0g)悬浮于水(285.0g)中并搅拌直至获得均匀的悬浮液。将均匀悬浮液投料至实验室规模的高压匀化设备中,该设备在60巴的压力下运行总共100个循环。在匀化步骤后,将悬浮液转移到保持容器中。将匀化的悬浮液投料至实验室规模的喷雾干燥器中,同时以5.7ml/min的进料速率并且在75℃的干燥温度(t out)下搅拌。
[0161]
分离的产物展示出与具有以下粒度分布的原料相同的xrpd:dv10=0.55μm;dv50=2.10μm;dv90=4.77μm;跨度=2.03,如图9所示。
[0162]
仪器参数
[0163]
nmr-核磁共振
[0164]
在bruker 300avance上,在300mhz(1h-nmr)和75mhz(
13
c-nmr)下记录1h和
13
c nmr波谱。
[0165]
ms-质谱法
[0166]
在quattro micro三重四级质谱仪(ireland)上进行ms实验,该三重四级质谱仪具有正离子模式(esi )的电喷射,120℃下的离子源,3.0kv的毛细管电压以及30v的源电压。
[0167]
hplc-高效液相色谱
[0168]
在以下条件下使用model alliance/2695和2487检测器(双λ)系统进行hplc分析:
[0169]
柱:waters symmetry shield rp18 4.6x150mm 3.5micra
[0170]
流速:0.8ml/min
[0171]
进样量:10ul
[0172]
温度:30℃
[0173]
溶剂a:h2o(0.1%tfa)
[0174]
溶剂b:ch3cn
[0175]
梯度洗脱方法如下:
[0176]
时间(分钟)流速(ml/min)流动相a(%)流动相b(%)0.010.8085.015.00.100.8085.015.036.000.8020.080.042.000.8020.080.042.100.8085.015.050.000.8085.015.0
[0177]
xrpd-x射线粉末衍射
[0178]
使用配备有铜源(cu/kα-)的panalytical x'pert pro x射线衍射系统来记录x射线粉末图案。
[0179]
dsc-差示扫描量热法
[0180]
在dsc q200上进行dsc实验,动态(ramp)10℃/min至350℃。
[0181]
tga-热重分析
[0182]
在tga q500上进行tga实验,动态10℃/min至350℃。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。