1.本发明涉及分子生物学技术领域,具体涉及一种花生组织单细胞核的提取方法及其应用。
背景技术:
2.植物单细胞测序实施相对比较困难,植物单细胞测序的难点在于分离具有活性的单个细胞,因为植物细胞具有细胞壁结构,尤其是高度木质化、纤维化、或成熟的组织。现阶段的方法主要是利用纤维素酶、果胶酶、离析酶的混合酶解液处理幼嫩的植物组织,通过降解细胞壁的纤维素、木质素、果胶后获得原生质体细胞,再利用原生质体细胞进行单细胞建库测序。然而,分离捕获单个不含细胞壁结构的细胞很难,这也极大地限制了植物单细胞测序技术的发展与应用。
3.单细胞转录组测序(single cell rna sequencing,scrna-seq)是在单个细胞内进行全部基因转录本的测序,不同于以往利用组织转录组测序(tissue bulk rna sequencing),该技术是在单细胞分辨率下鉴定基因表达的新方法。
4.单细胞核rna测序(single nucleus rna sequencing,snrna-seq)是在提取细胞核后通过微流控平台、nano-well平台标记细胞核再建库测序的方法。鉴于基因转录后信使rna会在细胞核中保存,单细胞核测序利用细胞核内存留的rna序列用以表征整个单细胞的基因表达量,该方法的优势在于不需要分离单细胞,适用于植物单细胞基因表达谱构建,以研究植物重要组织器官的发育进化。单细胞核rna测序通过使用单个细胞核,解决了样本解离引发的问题并提供了完善的技术方案,该技术可以针对大规模冰冻组织样本,以及高度木质化的植物组织样本开展单细胞测序,极大地扩展了单细胞测序技术在植物学研究中的应用。
5.因此,单细胞多组学技术(single cell multi-omics),即在同一个细胞内开展两种或者两种以上的测序,是植物单细胞测序重要的发展方向。
技术实现要素:
6.本发明的目的在于克服现有技术的不足之处而提供一种花生组织单细胞核的提取方法及其应用。
7.为实现上述目的,本发明采取的技术方案如下:
8.第一方面,本发明提供一种花生组织单细胞核的提取方法,包括以下步骤:
9.(1)取花生组织,粉碎至匀浆化且以不存在大片组织,得匀浆组织;
10.(2)向所得匀浆样组织加入核分离缓冲液,离心取上清,得上清液;
11.(3)取所得上清液过筛,得过筛上清液;
12.(4)取所得过筛上清液离心后弃上清,加入漂洗缓冲液重悬,即得单细胞核悬液。
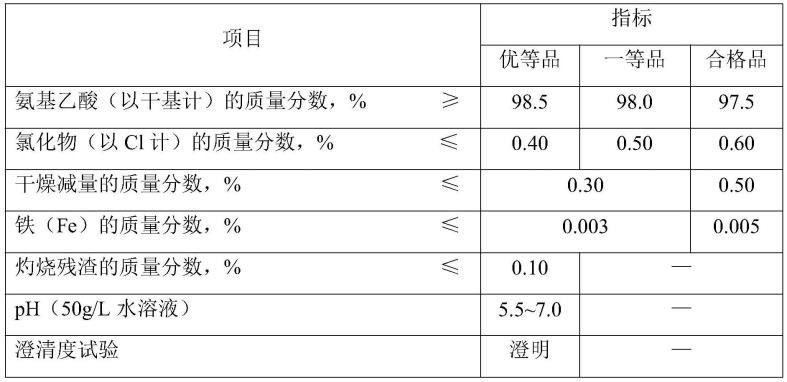
13.作为本发明所述的花生组织单细胞核的提取方法的优选实施方式,在步骤(2)中,所述核分离缓冲液包括终浓度为5%的dextran t40、0.4m的2m蔗糖、10mm的1m mgcl2、1mm
的1m二硫苏糖醇、2u/μl的rna酶抑制剂、0.1%的30%tritonx-100、100mm的1mtris hcl、0.2m的2-(n-吗啉代)乙磺酸、2m的kcl、1m的cacl2和1%蛋白酶抑制剂。
14.作为本发明所述的花生组织单细胞核的提取方法的优选实施方式,在步骤(2)中,所述离心采用200g~400g离心0.5min~1.5min。
15.作为本发明所述的花生组织单细胞核的提取方法的优选实施方式,在步骤(3)中,所述过筛时,先过65μm~75μm细胞筛,再过35μm~45μm细胞筛。
16.作为本发明所述的花生组织单细胞核的提取方法的优选实施方式,在步骤(4)中,所述漂洗缓冲液包括终浓度为10mm的pbs、1%的bsa、2u/μl的rna酶抑制剂、0.2m的2-(n-吗啉代)乙磺酸和2m的kcl。
17.作为本发明所述的花生组织单细胞核的提取方法的优选实施方式,在步骤(4)中所述离心采用1500g~2500g离心3min~8min。
18.第二方面,本发明提供了所述的提取方法制得的单细胞核悬液。
19.第三方面,本发明将所述的提取方法、所述单细胞核悬液在单细胞核转录组与单细胞核atac同胞建库中应用。
20.第四方面,本发明将所述的提取方法、所述单细胞核悬液在同胞单细胞核测序和单细胞核染色质开放性测序中应用。
21.第五方面,本发明将所述的提取方法、所述单细胞核悬液在植物基因特异性表达、鉴定细胞发育分化轨迹中应用。
22.与现有技术相比,本发明的有益效果为:
23.本发明开发了基于细胞核提取方法捕获单个细胞核,并利用单个细胞核开展同一细胞内的单细胞多组学研究,包括单细胞核转录组测序(snrna-seq)以及单细胞染色质开放性测序(snatac-seq),用单细胞核的基因表达数据表征整个单细胞的基因表达量,用单细胞核的atac测序结果表征单细胞的染色质开放性区间,即完成在同一单细胞核内同时获得转录组信息与染色质开放性区间信息,以此表征在单个细胞内同时获得两种组学信息,完成单细胞核表征的单细胞多组学测序。本发明促进花生分子生物学领域内的基因表达研究从单细胞水平继续深入到单个细胞器水平。
附图说明
24.图1为花生组织单细胞核粗提的荧光显微镜检测图。
25.图2为流式细胞仪荧光分选花生组织单细胞核悬液的荧光显图检测图。
26.图1和图2中,右图为荧光显微镜暗视野;中图为荧光显微镜明视野;作图为荧光显微镜暗视野和明视野叠加。
27.图3为分选荧光花生组织细胞核在流式细胞仪的上机参数。
28.图4为cell ranger软件分析获得的有效细胞数分析结果。
29.图5为cell ranger软件分析snrna-seq测序的细胞分群图之一。
30.图6为cell ranger软件分析snrna-seq测序的细胞分群图之二。
31.图7为snrna-seq测序结果分析流程示意图。
32.图8为snatac-seq测序结果分析流程示意图。
33.图9为花生叶片细胞单细胞核表征的单细胞多组学测序鉴定的亚细胞群。
34.图10为snrna-seq数据构建的花生叶片18个细胞群内的基因表达谱。
35.图11为snatac-seq数据构建的花生叶片18个细胞群内的染色质开放性图谱。
具体实施方式
36.为更好地说明本发明的目的、技术方案和优点,下面将结合具体实施例对本发明作进一步说明。本领域技术人员应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
37.实施例中所用的试验方法如无特殊说明,均为常规方法;所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
38.实施例1:花生组织单细胞核的提取
39.(1)配制溶液
40.配制核分离缓冲液,其配方如表1所示:
41.表1核分离缓冲液配方
42.试剂终浓度加入量dextran t405%0.5g2m蔗糖0.4m2ml1m mgcl210mm100μl1m二硫苏糖醇(dtt)1mm10μlrna酶抑制剂2u/μl50μl30%tritonx-1000.1%33μl1mtris hcl(ph7.4)100 mm1ml无rna酶水-定容至10ml
43.配制漂洗缓冲液wash buffer,其配方如表2所示:
44.表2 wash buffer配方
45.试剂终浓度加入量pbs10mm1mlbsa1%500μlrna酶抑制剂2u/μl50μl无rna酶水-定容至10ml
46.另配制改进核分离缓冲液和漂洗缓冲液wash buffer,具体如下:
47.改进核分离缓冲液与表1中配方不同之处在于,除表中试剂外,还加入了:2-(n-吗啉代)乙磺酸(mes,ph 5.7),终浓度0.2m,配制10ml中加入0.4625g;kcl,终浓度2m,配制10ml中加入1.491g;cacl2,终浓度1m,配制10ml中加入1.11g;蛋白酶抑制剂cocktail(100
×
),配制10ml中加入0.1ml。
48.改进wash buffer与表2中配方不同之处在于,除表中试剂外,还加入了:2-(n-吗啉代)乙磺酸(mes,ph 5.7),终浓度0.2m,配制10ml中加入0.4625g;kcl,终浓度2m,配制10ml中加入1.491g。
49.分别在离心管中配制好上述核分离缓冲液、wash buffer和改进核分离缓冲液、wash buffer,再分别用0.22μm过滤器过滤,即成。
50.(2)花生组织细胞核的提取方法具体如下:
51.1)用无菌剃须刀片将花生叶片切碎2分钟,重复2-3次,切至匀浆化且以不存在大片组织,得匀浆样品;
52.当花生样品组织较小时,可使用研钵和研杵在液氮中将冷冻组织小心地压碎成小块;
53.2)将匀浆样品小心转移至装有核分离缓冲液(nuclei isolation buffer,nib)或改进核分离缓冲液溶液的离心管中,以300g离心1min后取上清至新的离心管中,得上清液;
54.3)将上清液过70μm细胞筛至新50ml离心管,后再过40μm细胞筛至新15ml离心管中,得过筛上清液;
55.4)取过筛上清液以2000g离心5min后弃上清,加入wash buffer或改进wash buffer重悬,得单细胞核悬液,置于冰上备用。
56.(3)提取结果
57.采用改进前和改进后的核分离缓冲液和wash buffer所提取的花生细胞核数量统计结果如表3和表4所示:
58.表3提取细胞核数量
59.样品名称浓度(个/μl)体积(μl)细胞总数(个)细胞活性(%)检测结果备注改进后6601006600090c碎片率适中改进前3501003500085c碎片率高
60.表4提取细胞核dna质量检测
[0061][0062]
采用dapi将细胞核染色,荧光显微镜观察花生组织细胞核粗提结果,如图1所示。
[0063]
实施例2:分选细胞核
[0064]
具体步骤如下:
[0065]
(1)使用wash buffer将实施例1所得单细胞核悬液浓度调整至1000-2000核/μl;
[0066]
(2)流式细胞仪上样管预冷至4℃;取100μl过滤单细胞核悬液与900μl改进wash buffer混合转移到上样管中,作为非染色对照;
[0067]
使用70μm喷嘴,喷嘴的默认压力为20psi;根据fsc-a vs ssc-a,选择小颗粒gate,去掉大碎片(如图2所示);
[0068]
取单细胞核悬液中加入dapi染液,使终浓度为10μm;将染色的核悬液转移至流式上样管中。另取一流式上样管,加入1ml wash buffer,用于收集分选出的细胞核;根据dapi信号和核大小,对单细胞核进行分选,得分选单细胞核;
[0069]
(3)将分选单细胞核悬液在4℃、2000g离心5min后弃上清,加入wash buffer重悬;得重悬单细胞核悬液;
[0070]
(4)取10μl重悬单细胞核悬液加入终浓度10μm的dapi染液,使用dapi荧光通道检
查核质量和计数,将收集好的细胞核稀释至约1000核/μl;用于10xgenomics chromium微流控平台上机。
[0071]
实施例3:单细胞核转录组与单细胞核atac同胞建库
[0072]
1、试剂和仪器设备
[0073]
表5试剂和仪器设备
[0074][0075][0076]
建库所需的引物为:
[0077]
i7引物:接头序列ctgatcgtn,测序分析ctgatcgtnn;
[0078]
i5引物:接头序列atatgcgc,测序分析gcgcatat。
[0079]
2、实验方法
[0080]
(1)取实施例2分选的单细胞核悬液;
[0081]
(2)转座反应tn5酶处理剪切细胞核内染色质开放区间;
[0082]
a.按表6配制transposition mix溶液;
[0083]
表6 transposition mix
[0084]
transposition mix1
×
(μl)atac buffer7.0atac enzyme3.0
total10.0
[0085]
b.取5μl单细胞核悬液加入到10μl transposition mix中,吹打混匀;
[0086]
c.利用pcr仪按照表7中程序进行pcr反应,热盖温度50℃。得转座反应后单细胞核悬液。
[0087]
表7 pcr程序
[0088]
温度时间循环37℃60min-4℃
‑‑
[0089]
(3)制备gem和标记细胞
[0090]
a.将10
×
genomics chromium仪器启动芯片放置在芯片加上;
[0091]
b.按表8配制master mix溶液;
[0092]
表8 master mix
[0093]
master mix1
×
(μl)barcoding reagent49.5reducing agent b1.9template switch oligo1.1barcoding enzyme7.5total60.0
[0094]
c.将60μl master mix与15μl转座反应后单细胞核悬液混合,轻吹打匀;
[0095]
d.吸取70μl b中混合液、50μl gel bead、45μl partitioning oil加入芯片中;
[0096]
e.将芯片架放入chromium controller中,运行仪器;
[0097]
f.弹出芯片,吸取100μl转移到新pcr管;
[0098]
g.将pcr仪按表9中程序进行gem孵育,热盖温度50℃;
[0099]
表9 pcr程序
[0100][0101]
h.反应完全后,每个pcr管加入5μl quenching agent中止反应。得gem孵育单细胞核悬液。
[0102]
(4)gem孵育后的磁珠纯化
[0103]
1)dynabeads纯化
[0104]
a.室温下向每个gem孵育单细胞核悬液样品中添加125μl recovery agent,不可吹打或振荡混匀;
[0105]
b.从底部吸取并弃去125μl recovery agent/partitioning,注意不要吸到上清;
[0106]
c.按照表10配制dynabeads cleanup mix;
[0107]
表10 dynabeads cleanup mix
[0108]
dynabeads cleanup mix1
×
(μl)
cleanup buffer182dynabeads myone silane13reducing agent b5total200
[0109]
d.向每个样品加入200μl dynabeads cleanup mix,涡旋混匀,室温静置10min;
[0110]
e.孵育10min后,将ep管置于磁力架上,等待溶液变澄清,弃去上清;
[0111]
f.加入300μl 80%乙醇,静置30s,弃去乙醇;
[0112]
g.加入200μl 80%乙醇,静置30s,弃去乙醇;
[0113]
h.将ep管短暂离心后,放置到磁力架上,去除残留的乙醇溶液,室温干燥1min;
[0114]
i.按表11配置elution solution i;
[0115]
表11 elution solution i
[0116]
elution solution i10
×
(μl)buffer eb98010%tween 2010reducing agent b10total1000
[0117]
j.从磁力架上取下ep管,加入50.5μl elution solution i,吹打混匀,室温孵育1min;
[0118]
k.将ep管置于磁力架上,等待溶液变澄清;
[0119]
l.吸取50μl样品于新的ep管。得dynabeads纯化的单细胞核悬液。
[0120]
2)spriselect纯化
[0121]
a.取dynabeads纯化的单细胞核悬液,每个样本加入90μl spriselect reagent,充分吹打混匀,室温静置5min;
[0122]
b.短暂离心后,将ep管放置在磁力架上;
[0123]
c.待溶液澄清后,弃去上清;
[0124]
d.加入200μl 80%乙醇,静置30s,弃去乙醇;
[0125]
e.重复步骤d;
[0126]
f.短暂离心后,将ep管放到磁力架上,充分弃去所有乙醇;
[0127]
g.从磁力架上取下ep管,加入46.5μl buffer eb,吹打混匀,室温静置2min;
[0128]
h.短暂离线后,将ep管置于磁力架上;
[0129]
i.待到溶液澄清后,吸取46μl溶液转移到新的ep管中;得spriselect纯化的单细胞核悬液。
[0130]
(5)cdna/dsdna预扩增
[0131]
1)预扩增
[0132]
a.按表12配置pre-amplification mix
[0133]
表12 pre-amplification mix
[0134]
master mix1
×
(μl)pre-amp mix50pre-amp primers4
total54
[0135]
b.取spriselect纯化的单细胞核悬液,每个样本加入54μl pre-amplification mix,吹打混匀;
[0136]
c.利用pcr仪按表13中程序进行预扩增,热盖温度105℃。得预扩增样品。
[0137]
表13 pcr程序
[0138][0139][0140]
2)spriselect纯化
[0141]
a.取预扩增样品,每个样本加入160μl spriselect reagent,充分吹打混匀,室温静置5min;
[0142]
b.短暂离心后,将ep管放置于磁力架上;
[0143]
c.待溶液澄清后,弃去上清;
[0144]
d.加入300μl 80%乙醇,静置30s,弃去乙醇;
[0145]
e.加入200μl 80%乙醇,静置30s,弃去乙醇;
[0146]
f.短暂离心后,将ep管放置于磁力架上,弃去乙醇;
[0147]
g.从磁力架上取下ep管,加入160.5μl buffer eb,充分吹打混匀,室温静置2min;
[0148]
h.短暂离心后,将ep管放置于磁力架上;
[0149]
i.待到溶液澄清后,吸去160μl溶液转移到新的ep管;得纯化预扩增产物。
[0150]
(6)atac-seq文库构建
[0151]
1)样本索引pcr
[0152]
a.按表14配置pcr混合液;
[0153]
表14 pcr混合液
[0154]
master mix1
×
(μl)amp mix50si-pcr primer b7.5total57.5
[0155]
b.将57.5μl pcr混合液和40μl纯化预扩增产物混合,吹打混匀;
[0156]
c.在每个样本中加入2.5μl sample index n,set a,吹打混匀;
[0157]
d.将pcr仪按表15和表16中程序进行,热盖温度105℃;得样本索引pcr产物。
[0158]
表15 pcr程序
[0159][0160]
表16循环数对应表
[0161]
targeted nuclei recoverytotal cycles≤2,00092,001-6,00086001-100007
[0162]
2)pcr产物纯化
[0163]
a.取样本索引pcr产物,每个样本加入60μl spriselect reagent,充分吹打混匀,室温静置5min;
[0164]
b.短暂离心后,将ep管放置在磁力架上;
[0165]
c.待溶液澄清后,吸取150μl上清转移到新的ep管中;
[0166]
d.向收集的上清液中加入95μl spriselect reagent,吹打混匀,室温静置5min;短暂离心后,将ep管放置在磁力架上;
[0167]
e.待溶液澄清后,弃去上清;
[0168]
f.加入300μl 80%乙醇,静置30s,弃去乙醇;
[0169]
g.加入200μl 80%乙醇,静置30s,弃去乙醇;
[0170]
h.短暂离心后,将ep管放到磁力架上,充分弃去所有乙醇;
[0171]
i.从磁力架上取下ep管,加入20.5μl buffer eb,吹打混匀,室温静置2min;
[0172]
j.短暂离心后,将ep管置于磁力架上;
[0173]
k.待到溶液澄清后,吸去20μl溶液转移到新的ep管中;得纯化样本索引pcr产物。
[0174]
3)atac-seq文库质检
[0175]
利用dna 1000assay kit(agilent technologies安捷伦科技公司)或high sensitivity dna assay kit(agilent technologies安捷伦科技公司)进行文库质检。最后利用abi steponeplus real-time pcr system(life technologies)进行定量以及pooling,根据novaseq 6000的pe50模式上机测序。
[0176]
质检结果如表17所示:
[0177]
表17文库质检结果
[0178][0179]
(7)rna文库(基因表达文库)构建
[0180]
1)cdna扩增
[0181]
a.在冰上配制mix,如表18所示:
[0182]
表18 mix溶液
[0183]
cdna扩增mix1
×
(μl)amp mix50cdna primers15total65
[0184]
b.每个样品加入65μl cdna扩增mix和35μl纯化预扩增产物,混合,吹打混匀;
[0185]
d.将pcr仪按表19和表20中程序进行cdna扩增;得cdna扩增产物。
[0186]
表19 pcr程序
[0187][0188]
表20循环数对应表
[0189]
targeted nuclei recoverytotal cycles≤2,00092,001-6,00086001-100007
[0190]
2)cdna纯化
[0191]
a.每个样本加入60μl sprlselect reagent,充分吹打混匀,室温孵育5min;
[0192]
b.将ep管置于磁力架上,等待溶液变澄清;
[0193]
c.弃去上清;
[0194]
d.加入200μl新鲜配制的80%的乙醇溶液,等待30sec;
[0195]
e.吸弃乙醇;
[0196]
f.重复步骤e和f,总共洗涤2次;
[0197]
g.简短离心,置于磁力架上;
[0198]
h.去除残留的乙醇,室温干燥约约2min;
[0199]
i.从磁力架上取下ep管,马上加入加入40.5μl buffer eb,吹吸混匀15次;
[0200]
j.室温孵育2min;
[0201]
k.将ep管置于磁力架上,等待溶液变澄清;
[0202]
l.转移40μl样品于新的ep管;得纯化cdna扩增产物。
[0203]
3)片段化、末端修复加a
[0204]
a.冰上配制片段化混合液,如表21所示:
[0205]
表21片段化混合液
[0206]
fragmentation mix1
×
(μl)fragmentation buffer5fragmentation enzyme10total15
[0207]
b.取10μl纯化cdna扩增产物于ep管中;
[0208]
c.加入25μl buffer eb和15μl fragmentation mix,混匀离心;
[0209]
d.样品放入pcr仪进行反应32℃5min,65℃30min,4℃hold,65℃热盖;得片段化、末端修复加a扩增产物。
[0210]
4)片段筛选
[0211]
a.取片段化、末端修复加a扩增产物,每个样品加入30μl sprlselect,吹打混匀;
[0212]
b.室温孵育5min;
[0213]
c.将ep管置于磁力架上,等待溶液变澄清;
[0214]
d.转移75μl上清液到新的ep管;
[0215]
e.每个样品加入10μl sprlselect试剂,吹打混匀;
[0216]
f.室温孵育5min;
[0217]
g.将ep管置于磁力架上,等待溶液变澄清;
[0218]
h.吸弃80μl上清液,注意不要干扰到磁珠;
[0219]
i.加入125μl新鲜配制的80%的乙醇溶液,等待30sec;
[0220]
j.去除乙醇;
[0221]
k.重复步骤i和j,总共洗涤2次;
[0222]
l.简短离心,将ep管置于磁力架上,去除残留的乙醇;
[0223]
m.从磁力架上取下ep管,每个样品马上加入加入50.5μl buffer eb,混匀;
[0224]
n.室温孵育2min;
[0225]
o.将ep管置于磁力架上,等待溶液变澄清;
[0226]
p.转移50μl样品于新的ep管;得片段筛选扩增产物。
[0227]
5)接头连接
[0228]
a.按照以下表格配制接头连接mix,如表22所示;
[0229]
表22接头连接mix
[0230]
adaptor ligation mix1
×
(μl)ligation buffer20dna ligase10adaptor oligos20total50
[0231]
b.取片段筛选扩增产物,加入50μl adaptor ligation mix到样品中,混匀
[0232]
c.pcr仪运行以下程序:30℃热盖,20℃15min,4℃hold;得接头连接扩增产物。
[0233]
6)接头连接扩增产物纯化
[0234]
a.取接头连接扩增产物,每个样本加入80μl sprlselect reagent,充分吹打混匀,室温孵育5min;
[0235]
b.将ep管置于磁力架上,等待溶液变澄清;
[0236]
c.弃去上清;
[0237]
d.加入200μl新鲜配制的80%的乙醇溶液,等待30sec;
[0238]
e.吸弃乙醇;
[0239]
f.重复步骤e和f,总共洗涤2次;
[0240]
g.简短离心,置于磁力架上;
[0241]
h.去除残留的乙醇,室温干燥约约2min;
[0242]
i.从磁力架上取下ep管,马上加入加入30.5μl buffer eb,吹吸混匀15次;
[0243]
j.室温孵育2min;
[0244]
k.将ep管置于磁力架上,等待溶液变澄清;
[0245]
l.转移30μl样品于新的ep管;得纯化接头连接扩增产物。
[0246]
7)pcr扩增
[0247]
a.按照表23配制反应体系
[0248]
表23反应体系
[0249]
sample index pcr mix1
×
(μl)amp mix50si primer10total60
[0250]
b.加入60μl sample index pcr mix,10μl index,pcr反应;得扩增产物。
[0251]
8)扩增片段筛选
[0252]
a.取扩增产物,每个样品加入60μl sprlselect;
[0253]
b.室温孵育5min;
[0254]
c.将ep管置于磁力架上,等待溶液变澄清;
[0255]
d.转移150μl的上清液到新的ep管;
[0256]
e.每个样品加入20μl sprlselect;
[0257]
f.室温孵育5min;
[0258]
g.将ep管置于磁力架上,等待溶液变澄清;
[0259]
h.吸弃165μl上清液;
[0260]
i.加入200μl新鲜配制的80%的乙醇溶液,等待30sec;
[0261]
j.去除乙醇;
[0262]
k.重复步骤i和j,总共洗涤2次;
[0263]
l.简短离心,将ep管置于磁力架上,去除残留的乙醇;
[0264]
m.从磁力架上取下ep管,每个样品马上加入35.5μl buffer eb;
[0265]
n.室温孵育2min;
[0266]
o.将ep管置于磁力架上,等待溶液变澄清;
[0267]
p.转移35μl样品于新的ep管;得纯化扩增产物。
[0268]
9)基因表达文库质检及测序
[0269]
利用dna 1000assay kit(agilent technologies安捷伦科技公司)或high sensitivity dna assay kit(agilent technologies安捷伦科技公司)进行文库质检。最
后利用abi steponeplus real-time pcr system(life technologies)进行定量以及pooling,根据novaseq 6000的pe150模式上机测序。
[0270]
质检结果如表24所示:
[0271]
表24文库质检结果
[0272][0273]
实施例4:测序数据分析
[0274]
1、细胞质检
[0275]
将实施例2分选的单细胞核悬液与转座酶混合孵育,转座酶进入细胞核,优先在切割开放染色质区域,使dna片段化,并在dna片段的末端添加测序引物tcgcctta,得混合单细胞核悬液。
[0276]
2、10x genomics单细胞分离
[0277]
将含有barcode信息的凝胶珠与混合单细胞核悬液结合,然后被位于微流体系统中的油表面活性剂液滴包裹,形成gems(gel beads-in-emulsions)。gems流到储液器中并被收集,凝胶珠溶解,细胞核裂释放dna片段和mrna,并在片段末端加上标签。凝胶珠上一共有两种捕获序列,分别对应实施例3构建的atac-seq文库和基因表达文库。
[0278]
单细胞核转录组数据比对结果如表25所示:
[0279]
表25 snrna-seq测序结果质量分析表
[0280][0281]
单细胞核染色质开放性测序数据比对结果如表26所示:
[0282]
表26 snatac-seq测序结果质量分析表
[0283][0284]
3、测序文库构建
[0285]
对于scrna-seq文库,取实施例3制备的gem孵育单细胞核悬液,使用poly(dt)序列捕获pre-mrna和成熟mrna。随后,破裂gem结构,将所有cdna混合,pcr扩增获得足够构建文库的cdna量。随后,将cdna酶切打断为200~300bp左右的片段,然后经过末端修复、加a尾巴、测序接头p5、p7和sample index等常规二代测序文库构建步骤,最后进行pcr扩增得到标准测序文库。
[0286]
4、文库测序
[0287]
利用illumina测序平台的双端模式对scrna-seq单细胞转录组与scatac-seq单细胞染色质开放性文库进行高通量测序。对于scrna-seq文库,在read 1端,包含了16bp的
barcode信息和10bp的umi信息用于确定细胞和表达量;在read 2端,包含了cdna片段,用于参考基因组比对确定mrna所对应的基因。对于scatac-seq文库,在read1端,包含了16bp的barcode信息。在排除了10bp的i5和i7样本索引后,read1和read2端的剩余序列信息一起用于鉴定插入片段的信息。
[0288]
5、测序数据分析
[0289]
(1)序数据质控及基因表达量定量
[0290]
采用10x genomics官方分析软件cell ranger arc对scrna-seq文库和scatac-seq文库测序数据进行质量统计,并比对参考基因组,比对花生基因组数据库arahy.tifrunner.gnm1.kyv3(gcf_003086295.2)。结果如下:
[0291]
feature linkages detected:62,242;
[0292]
linked genes:12,472;
[0293]
linked peaks:67,977。
[0294]
cell ranger arc比对鉴定的数据结果如图4所示。
[0295]
cell ranger arc细胞聚类初步结果如图5和图6所示。
[0296]
(2)reads修剪
[0297]
对于实施例3制备的scrna-seq文库,在5'端去除30bp的tso序列,在在3'端去除polya序列;
[0298]
对于实施例3制备的scatac-seq文库,去除reads的接头和扩增引物序列。
[0299]
(3)snrna-seq数据
[0300]
illumina双末端测序结果中,read1包含16bp gemcode barcode(用于区分不同细胞)和10bp umi(unique molecular identifier,用于区分不同rna分子,一个mrna分子将会被一个umi标记);read2为cdna序列片段,用于进行基因组比对确定reads对应的基因。cell ranger arc首先对reads进行修剪,去除5’端的tso序列和序列和3’端的poly(a)序列。
[0301]
然后,调用star(spliced transcripts alignment to a reference)比对软件将将read2比对到参考基因组。依据gtf注释的结果,将比对上reads分类为外显子、内含或基因间区的分类为外显子、内含或基因间区的分类为外显子、内含或基因间区的分类为外显子、内含或基因间区reads:如一条reads有至少50%位于外显子区则属位于外显子reads,以此类推。如果reads比对到单一的外显子区域和一的外显子区域/多个非外显子区,则优先归类为外显子区且它的mapq被置为被置为255。
[0302]
cell ranger arc进一步将外显子reads比对回已注释的转录本上:首先,reads会被区分为正义链和反义链,正义链中比对到外显子、内含子和可变剪接形式reads会被进一步使用;其次,只有仅比对到一个基因上的reads会被最终用来进行umi定量。
[0303]
(4)snatac-seq数据
[0304]
illumina双末端测序结果中,read1端包含端包含16bp barcode(用于区分细胞)和插入片段信息,read2端包含插入片段另一信息。cell ranger arc会对reads进行预处理。
[0305]
首先对reads进行修剪,去除接头序列和引物。然后,调用bwa-mem将read pairs比对到参考基因组,根据reads比对到基因组的位置,构建插入片段,即fragments;同时,因为
bwa-mem需求reads长度至少是25bp,所以,低于该片段的reads会被标记为未必对上的reads。随后,cell ranger arc会对barcode序列进行校正,保留与已知的barcode序列一致的barcode,选择与已知的barcode序列差异小于等于2,综合barcode的reads丰度和错误碱基的测序质量进行评价,选择可信度》90%的barcode矫正为已知的barcode。最后,去除重复的片段序列,保留mapq》30的非线粒体,非嵌合体序列,完成fragments的过滤。
[0306]
(5)基因表达量定量
[0307]
cell ranger arc接着会过滤和校正umis。umi不允许是单寡聚链、不允许含有n、不允许含有质量值低于《10的碱基,否则会被过滤。如果某个umi与更高计数的umi只有一个错配且它们有相同的barcode和gene id,则它会被校正成较高计数的那个umi。只有效验证的reads才用于umi couting。
[0308]
将每个barcode的每个gene id对应的umi去重,计算unique umi的数量作为该细胞基因的表达量。
[0309]
(6)peak鉴定
[0310]
cell ranger arc会调用samtools将fragments正链末端位置 4bp,负链末端位置-5bp(因为转座反应占用9bp的区域),确定tn5转座酶的作用位点,即一次反应并保证转座酶的作用位点,并保证条fragments得到的两个转座反应间隔区域只与一条barcode相关。统合所有细胞的信息,计算出每个碱基发生的转座反应次数。随后对于每一个碱基,建立401bp的划窗,以zinba-like混合模型来生成一个平滑的信号窗。混合模型以负二项分布拟合噪音,一个几何分布零膨胀计数另信号值。最后设置比(odds-ratio)等于1/5为信号阈值,确定一个区域是否为peak信号,并将两个peak之间距离小于500bp的peak合并为一个peak作为最终的peak鉴定结果。
[0311]
(7)有效细胞数鉴定
[0312]
barcode的预处理会结合snrna-seq数据和snatac-seq数据,依照以下三个指标完成:
[0313]
(1)根据snatac-seq数据,滤fragments在peak的覆盖率低于fragments在基因组的覆盖率的barcodes。
[0314]
(2)根据snatac-seq数据,处理双凝胶珠barcode(两个凝胶珠和一细胞进入gem,致使一个细胞的数据被拆分为两组信息)。挑选barcodes(b1,b2),如果b1和b2之间fragments的邻近程度高于自身(b1与b1和b2与b2)的邻近程度,则认为b1和b2是来源于同一个细胞。
[0315]
(3)结合两组学数据,保留在中都至少检测到一条umi或一条fragments的barcodes信息。
[0316]
在完成以上步骤后,就可结合两组学的数据,进行有效细胞筛选,具体为:
[0317]
首先将过滤barcode信息绘制在一张二维图像中,横坐标为snatac-seq得到的每个barcode包含转座次数,纵坐标为snrna-seq得到的每个barcode包含的umi数量,具有相同横纵坐标信息的barcode被压缩为一个点。在这的图形基础上使用被压缩为一个点。然后,在这的图形基础上使用ordmag算法定义细胞和非的划分阈值,这个算法对snrna-seq snatac-seq使用。随后通过k-means算法,以ordmag算法得到的细胞和非群体为中心,设置k=2,对细胞和非细胞进行聚类,以此优化划分阈值。将细胞进行聚类,以此优化划分阈值。
最后将k-means分类结果应用于所有过滤后barcodes中,形成最终鉴定的有效细胞群体。
[0318]
试验例:
[0319]
1、分别采集花生幼苗生长到3,5,7天,取幼嫩的叶片按照实施例1的方法提取细胞核粗提液,dapi染色后,用流式细胞仪荧光分选,获得纯净的细胞核,利用tn5酶处理后,获得细胞核悬液。
[0320]
2、取tn5酶处理的细胞核悬液,反向包裹gem凝胶磁珠,形成单细胞核与单个磁珠的油滴小泡,收集凝胶磁珠小泡,通过裂解释放mrna以及染色质dna片段,与接头引物融合后,分别建库测序。
[0321]
3、建库测序原始数据,利用10x genomics官方分析软件cell ranger arc对原始数据进行数据过滤、鉴定回收细胞,最终得到各细胞的基因表达图谱与染色质开放图谱。获得了花生叶片单细胞核表征的单细胞基因表达谱与染色质开放性表达谱,共计5930个单细胞,两个数据整合分析将叶片细胞划分为18个亚细胞群,用以鉴定细胞类型,如图9所示。构建花生叶片5930个细胞的单细胞基因表达图谱与单细胞染色质开放性图谱,分别为叶肉细胞、维管细胞、保卫细胞、表皮细胞、原基细胞与薄壁细胞,为植物基因特异性表达、鉴定细胞发育分化轨迹提供基础。在单细胞层面研究细胞分化发育、基因表达最终对整个组织的生长发育产生的影响。
[0322]
snrna-seq单细胞基因表达图谱如图10所示;snatac-seq单细胞染色质开放性图谱如图11所示。
[0323]
具体实验分析流程为:
[0324]
10x genomics atac rna联合试剂,在分离细胞核并保证细胞核核膜完整、活性良好后,首先利用转座酶混合液孵育细胞核悬液,优先在切割开放染色质区域使dna片段化,并在dna片段的末端添加测序引物。之后,将带有barcode标签和捕获序列的凝胶珠和单个细胞核包裹在油滴中,形成gems(gel-beads in emulsion)。随后,凝胶珠溶解释放捕获序列,细胞核破裂释放dna片段、pre-mrna和成熟mrna,并在片段末端加上标签。凝胶珠上一共有两种捕获序列,分别对应atac-seq文库和基因表达文库。
[0325]
一种是poly(dt)序列,用于捕获pre-mrna和成熟mrna,对应基因表达文库。添加标签后,破裂gem结构,将所有cdna混合,pcr扩增获得足够构建文库的cdna量。随后,将cdna酶切打断为200~300bp左右的片段,然后经过末端修复、加a尾、测序接头p5、p7和sample index等常规二代测序文库构建步骤,最后进行pcr扩增得到标准测序文库。
[0326]
一种是spacer序列,用于捕获dna片段,对应atac-seq文库。添加标签后,破裂gem结构,将所有dsdna混合,pcr扩增获得足够构建的dna量。随后,通过pcr增加测序接头p7、sample index添加到dna片段上,构建成标准测序文库。
[0327]
基因表达图谱分析流程snrna-seq分析流程如图7所示。
[0328]
染色质开放性图谱snatac-seq分析流程如图8所示。
[0329]
用单细胞核的基因表达数据表征整个单细胞的基因表达量,用单细胞核的atac测序结果表征单细胞的染色质开放性区间,通过barcode整合两个数据库信息,即完成在同一单细胞核内同时获得转录组信息与染色质开放性区间信息,以此表征在单个细胞内同时获得两种组学信息,完成单细胞核表征的单细胞多组学测序。
[0330]
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保
护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。