一种吲哚生物碱luteoride a的首次全合成工艺
技术领域
1.本发明涉及luteoridea合成技术领域,具体为一种吲哚生物碱luteoridea的首次全合成工艺。
背景技术:
2.目前尚无在无保护基操作下在吲哚7位直接引入异戊二烯基团的先例,传统方法在引入异戊二烯基团的过程中具有一定的局限性,通常需要苛刻的反应条件强酸强碱等、高毒性试剂(三甲基硅烷化重氮甲烷、氰化亚铜、2-甲基-1-丁烯-3-炔等)、高度易燃试剂(正丁基锂、三正丁基氢锡、儿茶酚硼烷等)以及昂贵试剂(三甲基硅烷化重氮甲烷、儿萘酚硼烷、2-甲基-1-丁烯-3-炔等)的使用。原子经济性不好,制备成本高,总产率低,并且目前还没有关于吲哚生物碱luteoridea合成的相关报道。
3.基于此,本发明设计了一种吲哚生物碱luteoridea的首次全合成工艺,以解决上述问题。
技术实现要素:
4.本发明的目的在于提供一种吲哚生物碱luteoridea的首次全合成工艺,以解决上述背景技术中提出的问题。
5.为实现上述目的,本发明提供如下技术方案:一种吲哚生物碱luteoridea的首次全合成工艺,所述吲哚生物碱luteoridea的首次全合成工艺包括以下步骤:
6.s1:底物4为起始物料为原料与盐酸羟胺发生羟胺化反应以87%的收率得到化合物5;具体为:在氮气保护下于干燥反应瓶中加入335ml chc13,和221mlch3oh,室温下加入底物417.7ml(165.8mmol),搅拌均匀,然后加入10.7g盐酸经胺(165.8mmol);混合物在室温下搅拌一夜,然后在30℃旋转蒸发浓缩混合物除去大部分溶剂,有少量溶剂时残留停止;将残渣溶于300ml二氯甲烷中,依次用0.1n的30ml hc1、230ml水和30ml饱和的氯化钠溶液洗涤,合并有机相用无水硫酸钠干燥,过滤,浓缩后得到28.3g白色晶体化合物5(产率87%);不需要进一步的纯化。
[0007][0008]
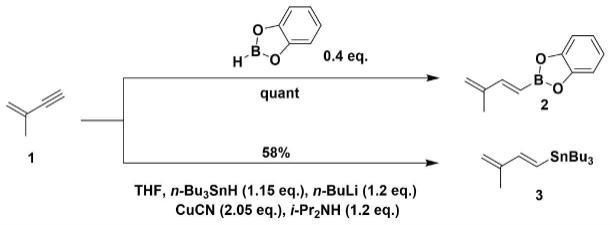
s2:用过量的起始原料2-甲基-1-丁烯-3-炔1加入儿萘酚硼烷得到化合物2;
[0009]
在用2-甲基-1-丁烯-3-炔1中加入化合物氰化亚铜以及易燃化合物正丁基锂、三正丁基氢锡得到化合物3。
[0010]
制备异戊二烯基化合物
[0011][0012]
化合物5与7-溴吲哚底物6发生杂原子d-a反应后开环并芳构化以82%的收率得到化合物7,具体为:在氮气保护下,10g化合物2(51mmol)用110ml二氯甲烷溶解后滴加到用110ml二氯甲烷溶解和10.8g na2co3(102mmol)处理的20g 7-溴吲哚3(102mmol)中,混合物在室温下搅拌10h,然后用200ml二氯甲烷萃取,依次用220ml水和20ml饱和氯化钠溶液洗涤,合并有机相后用无水硫酸钠干燥,过滤,然后在30℃旋转蒸发浓缩除去溶剂得到化合物2,残留物经用硅胶柱层析纯化(石油醚:乙酸乙酯=5:1至3:1)得13g化合物4(产率82%)。
[0013][0014]
s3:化合物7与2-甲基-3-丁烯-2-醇8在钯催化剂参与下发生heck反应并原位消除三级羟基以94%的产率得到luteoridea;具体为:在氮气保护下,在封管中先后投入1.12g化合物4(3.6mol)、64.8mg催化剂(0.288mmo1)、219.6mg三(邻甲基苯基)膦(0.72mmol)、79.2mg bht(0.36mmol),dmf 19.2ml、772μl n-甲基二环已基胺(3.6mmol)及1.7ml 2-甲基-3-丁烯-2-醇5(54mmol),混合均匀后于油浴115
°
中反应6小时,冷却后用用10ml碳酸氢钠溶液搅拌5分钟后,硅胶过滤不溶物并用乙酸乙酯150ml洗涤滤饼,滤液一次用(5
×
30ml)水和30ml饱和氯化钠溶液洗涤,合并有机相后用无水硫酸钠干燥,过滤,然后在30℃旋转蒸发浓缩除去溶剂;残留物经用硅胶柱层析纯化(石油醚:乙酸乙酯=5:1-1:1)得978mg luteoride a(产率92%)。
[0015][0016]
作为本发明的进一步方案,所述步骤一中底物4为溴代丙酮酸甲酯、溴代丙酮酸乙酯、溴代丙酮酸乙酯、氯代丙酮酸甲酯、氯代丙酮酸乙酯、氯代丙酮酸丙酯、碘代丙酮酸甲酯、碘代丙酮酸乙酯或碘代丙酮酸丙酯。
[0017]
作为本发明的进一步方案,所述步骤三中催化剂为醋酸钯、氯化钯、四三苯基膦钯、三(二亚基丙酮)二钯-氯仿加合物、三(二亚基丙酮)二钯、三氟乙酸钯、二(三叔丁基膦)钯、钯碳或者不添加催化剂。
[0018]
作为本发明的进一步方案,所述步骤二中底物6为7-溴吲哚、4-溴吲哚、5-溴吲哚或6-溴吲哚。
[0019]
与现有技术相比,本发明的有益效果是:
[0020]
1.本发明路线首次在吲哚7位引入异戊二烯基团,不仅避免了毒性试剂(三甲基硅烷化重氮甲烷、氰化亚铜、2-甲基-1-丁烯-3-炔等)、高度易燃试剂(正丁基锂、三正丁基氢锡等)以及昂贵试剂(三甲基硅烷化重氮甲烷、儿萘酚硼烷、2-甲基-1-丁烯-3-炔等)的使用,而且还使用无保护基操作,具有氧化还原经济性以及原子经济性;并且实现了吲哚生物碱luteoridea的首次全合成;总体来说,该专利合成方法具有路线简捷,原料简单易得,制备成本较低,产品收率高,操作简便等优点。
附图说明
[0021]
图1为本发明底物5的核磁共振氢谱示意图;
[0022]
图2为本发明底物5的核磁共振碳谱示意图;
[0023]
图3为本发明底物7的核磁共振氢谱示意图;
[0024]
图4为本发明底物7的核磁共振碳谱示意图;
[0025]
图5为本发明luteoridea的核磁共振氢谱示意图;
[0026]
图6为本发明luteoridea的核磁共振碳谱示意图。
具体实施方式
[0027]
请参阅图1-6,本发明提供一种技术方案:一种吲哚生物碱luteoridea的首次全合成工艺,所述吲哚生物碱luteoridea的首次全合成工艺包括以下步骤:
[0028]
s1:底物4为起始物料为原料与盐酸羟胺发生羟胺化反应以87%的收率得到化合物5;具体为:在氮气保护下于干燥反应瓶中加入335ml chc13,和221mlch3oh,室温下加入底物417.7ml(165.8mmol),搅拌均匀,然后加入10.7g盐酸经胺(165.8mmol);混合物在室温下搅拌一夜,然后在30℃旋转蒸发浓缩混合物除去大部分溶剂,有少量溶剂时残留停止;将残渣溶于300ml二氯甲烷中,依次用0.1n的30ml hc1、230ml水和30ml饱和的氯化钠溶液洗涤,合并有机相用无水硫酸钠干燥,过滤,浓缩后得到28.3g白色晶体化合物5(产率87%);不需要进一步的纯化。
[0029][0030]
白色固体,总收率87%,1h nmr(400mhz,acetone-d6)δ4.39(d,2h),3.82(s,3h);
13
c nmr(101mhz,acetone-d6)δ162.73,147.57,51.92,15.97.
[0031]
s2:用过量的起始原料2-甲基-1-丁烯-3-炔1加入儿萘酚硼烷得到化合物2;
[0032]
再用2-甲基-1-丁烯-3-炔1中加入化合物氰化亚铜以及易燃化合物正丁基锂、三正丁基氢锡得到化合物3。
[0033]
制备异戊二烯基化合物
[0034][0035]
化合物5与7-溴吲哚底物6发生杂原子d-a反应后开环并芳构化以82%的收率得到化合物7,具体为:在氮气保护下,10g化合物2(51mmol)用110ml二氯甲烷溶解后滴加到用110ml二氯甲烷溶解和10.8g na2co3(102mmol)处理的20g 7-溴吲哚3(102mmol)中,混合物在室温下搅拌10h,然后用200ml二氯甲烷萃取,依次用220ml水和20ml饱和氯化钠溶液洗涤,合并有机相后用无水硫酸钠干燥,过滤,然后在30℃旋转蒸发浓缩除去溶剂得到化合物2,残留物经用硅胶柱层析纯化(石油醚:乙酸乙酯=5:1至3:1)得13g化合物4(产率82%)。
[0036][0037]
固体,总收率82%,1h nmr(400mhz,cdcl3)δ9.47(s,1h),8.22(s,1h),7.72(dt,j=7.9,0.8hz,1h),7.34(dd,j=7.7,0.8hz,1h),7.21(d,j=2.4hz,1h),7.01(t,j=7.8hz,1h),4.09(d,j=0.9hz,2h),3.82(s,3h);
13
c nmr(101mhz,cdcl3)δ163.78,151.23,134.55,128.39,124.42,124.17,120.77,118.54,110.77,104.67,52.81,20.45.
[0038]
s3:化合物7与2-甲基-3-丁烯-2-醇8在钯催化剂参与下发生heck反应并原位消除三级羟基以94%的产率得到luteoridea;具体为:在氮气保护下,在封管中先后投入1.12g化合物4(3.6mol)、64.8mg催化剂(0.288mmo1)、219.6mg三(邻甲基苯基)膦(0.72mmol)、79.2mg bht(0.36mmol),dmf 19.2ml、772μl n-甲基二环已基胺(3.6mmol)及1.7ml 2-甲基-3-丁烯-2-醇5(54mmol),混合均匀后于油浴115
°
中反应6小时,冷却后用用10ml碳酸氢钠溶液搅拌5分钟后,硅胶过滤不溶物并用乙酸乙酯150ml洗涤滤饼,滤液一次用(5
×
30ml)水和30ml饱和氯化钠溶液洗涤,合并有机相后用无水硫酸钠干燥,过滤,然后在30℃旋转蒸发浓缩除去溶剂;残留物经用硅胶柱层析纯化(石油醚:乙酸乙酯=5:1-1:1)得978mg luteoride a(产率92%)。
[0039][0040]
固体,总收率92%,1h nmr(400mhz,acetone-d6)δ11.48(s,1h),10.44(s,1h),7.66(d,j=7.9hz,1h),7.37(d,j=7.4hz,1h),7.19(d,j=2.3hz,1h),7.07(d,j=4.0hz,2h),7.03(d,j=7.7hz,1h),5.15(d,j=2.3hz,1h),5.06(dt,j=2.9,1.4hz,1h),4.06(s,
2h),3.70(s,3h);
13
c nmr(101mhz,acetone-d6)δ164.40,151.07,142.58,134.45,131.54,128.19,124.55,123.88,121.24,119.19,118.50,118.36,116.37,109.74,51.43,19.98,17.88;ir(kbr):3332,3050,2944,2346,1705,1438,1340,1206,1106,1013,894,799,728,670,466cm-1
;hrms(esi)m/z calcd for c
17h17
n2o3[m-h]
297.1246,found297.1245.
[0041]
作为本发明的进一步方案,所述步骤一中底物4为溴代丙酮酸甲酯、溴代丙酮酸乙酯、溴代丙酮酸乙酯、氯代丙酮酸甲酯、氯代丙酮酸乙酯、氯代丙酮酸丙酯、碘代丙酮酸甲酯、碘代丙酮酸乙酯或碘代丙酮酸丙酯。
[0042]
作为本发明的进一步方案,所述步骤三中催化剂为醋酸钯、氯化钯、四三苯基膦钯、三(二亚基丙酮)二钯-氯仿加合物、三(二亚基丙酮)二钯、三氟乙酸钯、二(三叔丁基膦)钯、钯碳或者不添加催化剂。
[0043]
作为本发明的进一步方案,所述步骤二中底物6为7-溴吲哚、4-溴吲哚、5-溴吲哚或6-溴吲哚。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
