一种基于crispr-cas9系统进行枯草芽孢杆菌基因组编辑的方法及其应用
技术领域
1.本发明涉及芽孢杆菌基因组编辑领域,更具体地涉及一种基于crispr-cas9系统进行枯草芽孢杆菌基因组编辑的方法及其应用。
背景技术:
2.枯草杆菌遗传操作的传统策略是基于同源重组。这些基因组编辑方法包括使用抗生素耐药标记、反选择标记和cre/loxp系统。近年来,起源于细菌适应性免疫系统的crispr-cas9系统已被应用于许多原核和真核生物的遗传操作。crispr-cas9系统效率高,使用省时,修饰后的生物体没有标记。在枯草芽孢杆菌中已经使用了三种不同的crispr-cas9系统:单质粒系统、双质粒系统和染色体系统。在单导rna(sgrna)的辅助下,cas9蛋白可以在枯草杆菌基因组的靶部位引入双链断裂(dsb)。然后,同源修复模板引导同源重组,从而进行基因的更改,敲除或插入。
3.近些年来,人们不断对crispr-cas9系统进行研究。例如,基于簇状规则间隔短回文重复序列/相关蛋白9(crispr/cas9)的碱基编辑技术是crispr技术家族的最新成员。与传统的crispr/cas9技术相比,它不依赖于dna双链断裂和同源重组,可以更快、更简单地实现基因失活和点突变。有课题组首次利用crispr/dcas9(化脓链霉菌cas9的全核酸酶缺陷突变体)和激活诱导型胞苷脱氨酶(aid),建立了枯草芽孢杆菌基因组编辑的碱基编辑方法。该方法实现了3个和4个位点的同时编辑,编辑效率分别达到100%和50%。
4.seca的膜结合涉及带负电荷的磷脂和secy的胞质环。seca对secy的亲和力远高于其对细胞质中前蛋白的亲和力,因此seca被认为是secyeg转运子的可溶性受体亚基。蛋白质分泌前需要seca与膜蛋白即分泌通道蛋白形成一个稳定的复合结构,以此来保证蛋白质的稳定分泌。seca/前蛋白复合物一旦到达内膜,在借助seca重复进行atp水解释放能量后,分泌蛋白会进入secyeg转运蛋白并借助secdf拉拽作用完成分泌。
5.secyeg是镶嵌在细胞膜上的异三聚体蛋白质复合物,它是蛋白质易位的主要参与者,并充当胞内大分子伴侣蛋白seca停靠的膜通道。secyeg同样可以提供能量来使未完全折叠的前蛋白通过其水性内部分泌。因此,seca与膜复合物secyeg形成稳定的全转位酶可能会有利于蛋白的分泌。
6.当seca与蛋白质前体结合之后,seca/前体蛋白复合体会靶向细胞膜上的分泌通道并与之结合,亚基secy是通道的主体部分。seca/前体蛋白复合体第一时间靶向分泌通道后,与secy进行结合,形成稳定的分泌复合体。此时,seca上会存在氨基酸残基(红色圈中的氨基酸残基)与磷脂进行相互作用。这种相互作用是通过两个带电区域发挥作用的,一个包括seca的hsd中的r553、r576和k583残基,另一个包括螺旋翼域(hwd)中的r645和e646残基。seca与secy的复合结构如图1所示。其中,红色圈中的氨基酸是seca的hwd中的e646残基。
7.当atp与seca结合并在前体蛋白开始被释放时,前体蛋白的信号序列n末端会形成一个a螺旋结构,该结构极有可能被seca的底物结合钳捕捉,并被输送至由secy组分形成的
通道中。
8.在枯草杆菌中,组件seca的角色,结构及作用机制也逐渐被解析,为基于结构认知的理性改造提供了基础。kakeshita等人发现seca的c末端结构域的删除促进了两种不同的异源蛋白耐热碱性纤维素酶和人α干扰素的分泌;diao等人将枯草杆菌中seca的c末端替换为大肠杆菌seca的c末端结构域后并同时过表达secb蛋白,提高了分泌不佳的突变麦芽糖结合蛋白(male11)和碱性磷酸酶(phoa)的分泌能力,这说明了基于结构对枯草分泌组件的理性改造能够有效改善异源蛋白的分泌能力。
9.然而,现有技术的缺点在于:1)现有的枯草芽孢杆菌基因组编辑技术基因组点突变编辑效率低,操作复杂,对crispr系统要求高;2)现有的提高异源蛋白在枯草芽孢杆菌中分泌能力的技术,如信号肽筛选及修饰,工作量大;3)现有的分泌途径改造盲目性大,针对性不强,分子操作复杂。
技术实现要素:
10.本发明的目的是提供一种基于crispr-cas9系统进行芽孢杆菌基因组编辑的方法及其应用,从而解决现有技术中枯草芽孢杆菌基因组编辑技术基因组点突变编辑效率低,操作复杂,分泌途径改造盲目性大的问题。
11.根据本发明的第一方面,提供一种基于crispr-cas9系统进行枯草芽孢杆菌基因组编辑的方法,所述方法包括以下步骤:s1:crrna序列的获得:根据需要靶向枯草芽孢杆菌基因组的靶基因序列设计获得crrna序列;s2:sgrna表达质粒的构建:将所述crrna序列与tracrrna融合组成sgrna,获得sgrna表达质粒;s3:crrna同义突变设计:基于密码子的简并性,将所述crrna序列同义突变成t20-crrna,完成枯草芽孢杆菌基因组靶基因crrna序列的替换,以防止sgrna的靶向识别;s4:crispr/cas9质粒的构建:设计引物对靶基因的上、下游同源臂进行扩增,其中,上游同源臂的反向引物含有所述t20-crrna序列,下游同源臂的正向引物含有所述t20-crrna序列,通过扩增获得纯净的上、下游同源臂dna片段,并连接成融合片段,即为donor dna质粒,然后将所述donor dna质粒与所述sgrna表达质粒进行融合,最终获得crispr/cas9质粒;s5:枯草芽孢杆菌基因组编辑:将步骤s4构建完成的crispr/cas9质粒转化枯草芽孢杆菌感受态细胞中,丢去质粒,即可完成枯草芽孢杆菌基因组编辑;s6:验证编辑结果;设计引物,扩增编辑后宿主的靶基因,对pcr产物测序进行验证。
12.进一步地,该方法该包括步骤s7:验证编辑效率:将t20-crrna序列插入至tracrrna,获得改造后的crispr/cas9质粒,接着将基因组野生型靶基因作为同源模板用于修复双链的断裂,对t20宿主基因组的靶基因进行编辑,完成基因组编辑后,宿主进行丢去质粒,随机选取多个菌落进行靶基因扩增,并送去测序,比对测序结果,记录编辑成功菌落个数,以此计算编辑成功率。
13.步骤s1包括:从ncbi网站上查询靶基因序列,将靶基因输入crrna设计网站中,以此获得网站推荐的crrna序列。
14.所述枯草芽孢杆菌基因组的靶基因可选自枯草芽孢杆菌基因组,比如seca基因,secy基因,sece基因,secg基因,secd基因,secf基因等。
15.步骤s2包括:以pjoe8999质粒为例,将pjoe8999质粒作为模板,通过将步骤s1设计的所述crrna序列设计在引物的5’端,使用引物进行pcr扩增,回收产物使用无缝克隆酶连
接成环状质粒,即,sgrna表达质粒。
16.步骤s4包括:首先,使用引物对所述靶基因的上、下游同源臂进行pcr,获得上、下游同源臂dna片段;接着,以所述上、下游同源臂dna片段为模板,进行融合pcr,获得融合片段;对所述融合片段和sgrna表达质粒进行融合,转化dh5a,涂布含有卡那抗生素抗性的lb平板上,过夜培养,菌落pcr验证正确,质粒抽提,获得crispr/cas9质粒。
17.步骤s5包括:将步骤s4构建完成的质粒转化枯草芽孢杆菌感受态细胞中,将其涂布在含有5μg/ml卡那霉素和0.2%甘露糖的lb平板上培养,在平板上标记出已生长出来的菌落,培养结束后,将标记的菌落点样在不含抗生素的lb平板上,在50℃条件下培养16-18h,丢去质粒,最后将菌落对应点样于含有50μg/ml卡那霉素lb平板上过夜培养,验证质粒是否丢失。
18.步骤s6包括:在确认基因组编辑完成,宿主质粒完全丢失后,扩增编辑后宿主的靶基因,并进行琼脂糖凝胶电泳验证pcr结果,获得目的条带后对pcr产物进行测序,结果正确则将此宿主命名为t20。
19.根据本发明的一种具体实施方式,步骤s1包括:从ncbi网站上查询seca基因序列,将seca基因输入crrna设计网站中,以此获得网站推荐的crrna序列,如seq id no.13所示。步骤s2包括:以pjoe8999质粒为模板,通过将步骤s1设计的所述crrna序列设计在引物的5’端,使用引物secagrna-f和secagrna-r进行pcr扩增,回收产物使用无缝克隆酶连接成环状质粒,即,sgrna表达质粒骨架pjoe8999-secagrna构建完成,所述引物secagrna-f和secagrna-r的序列如seq id no.1-2所示。s3:crrna同义突变设计:基于密码子的简并性,将crrna序列同义突变成t20-crrna,如seq id no.14,完成枯草芽孢杆菌基因组靶基因crrna序列的替换,以防止sgrna的靶向识别。步骤s4包括:首先,使用引物seca-sfi
ⅰ‑
f与t20-r对seca上游同源臂进行pcr,引物seca-sfi
ⅰ‑
r与t20-f对下游同源臂进行pcr,即可获得纯净的上、下游同源臂dna片段;接着,以所述上、下游同源臂dna片段为模板,seca-sfi
ⅰ‑
f/seca-sfi
ⅰ‑
r为引物,使用kod one进行融合pcr,获得融合片段;然后,对所述融合片段和pjoe8999-secagrna质粒进行sfiⅰ酶切,使其具有粘性末端,随后使用t4连接酶进行连接,转化dh5a,涂布含有卡那抗生素抗性的lb平板上,过夜培养,菌落pcr验证正确,质粒抽提,获得质粒pjoe8999-t20secagrna;其中,引物seca-sfi
ⅰ‑
f与seca-sfi
ⅰ‑
r的序列如seq id no.3-4所示,引物t20-f与t20-r的序列如seq id no.5-6所示。步骤s5包括:将步骤s4构建完成的质粒pjoe8999-t20secagrna转化枯草芽孢杆菌感受态细胞中,将其涂布在含有5μg/ml卡那霉素和0.2%甘露糖的lb平板上培养,在平板上标记出已生长出来的菌落,培养结束后,将标记的菌落点样在不含抗生素的lb平板上,在50℃条件下培养16-18h,丢去质粒,最后将菌落对应点样于含有50μg/ml卡那霉素lb平板上过夜培养,验证质粒是否丢失。步骤s6包括:在确认基因组编辑完成,宿主质粒完全丢失后,以seca-sfi
ⅰ‑
f/seca-sfi
ⅰ‑
r为引物,扩增编辑后宿主的seca基因,并进行琼脂糖凝胶电泳验证pcr结果,获得目的条带后对pcr产物进行测序,结果正确则将此宿主命名为t20。
20.根据本发明的第二方面,提供一种如上面所述的基于crispr-cas9系统进行枯草芽孢杆菌基因组编辑的方法在对枯草芽孢杆菌进行分泌途径改造中的应用。
21.根据本发明的一个实施方式,采用如上面所述的基于crispr-cas9系统进行枯草芽孢杆菌基因组编辑的方法,将t20宿主基因组seca上的646位点谷氨酸替换成酪氨酸y或
异亮氨酸i,实现对t20宿主的基因点突变,分别构造出蛋白分泌能力提高的宿主e646y、e646i,实现了对枯草芽孢杆菌的分泌途径改造。
22.应当理解的是,本发明并不仅限于将t20宿主基因组seca上的e646谷氨酸替换成酪氨酸y或异亮氨酸i,实际上,对于seca上e646位点的改造还包括可以将异亮氨酸i和酪氨酸y换成其他氨基酸进行研究。
23.还应当理解的是,虽然本发明重点以seca基因为例对该基因组编辑方法进行阐述,但是实际上本发明的基因组编辑方法并不仅限于枯草芽孢杆菌基因组的seca基因,所述枯草芽孢杆菌基因组的靶基因还可以选自枯草芽孢杆菌基因组其他基因。
24.本发明的关键发明点在于,基于最基础的crispr-cas9系统能够高效的对枯草芽孢杆菌基因组进行定点突变,从而对枯草芽孢杆菌进行分泌途径改造,提高了枯草芽孢杆菌对碱性蛋白酶的分泌能力,首次通过理性设计改造枯草芽孢杆菌分泌途径关键组分seca提高异源蛋白分泌效率。
25.其次,由于密码子具有简并性,因此可以对crrna进行同义突变,改变核酸序列,但是不改变氨基酸序列,不影响seca的功能。因此,在构建用于同源重组的编辑模板时,将原有crrna序列同义突变,突变后的crrna序列称为t20-crrna,将原crrna序列尽最大可能变为与原序列不同的t20-crrna序列,以防止修复模板被sgrna的靶向识别。
26.再次,本发明的另一发明点还在于,考虑到seca上的e646会与疏水性的磷脂进行相互作用,本发明采用疏水性最强的带有脂肪族侧链的异亮氨酸i,和亲水性的带有芳香族侧链的酪氨酸y对带有较弱负电性的谷氨酸e进行替换,旨在使seca与膜蛋白(膜蛋白形成了膜上的分泌通道)结合更稳定,以提高蛋白分泌能力。
27.根据本发明提供的一种基于crispr-cas9系统进行芽孢杆菌基因组编辑的方法以及在对枯草芽孢杆菌进行分泌途径改造中的应用,其相对现有技术具有以下优越性:
28.1)由于cas9在切割基因组双链dna的时候需要sgrna的靶向作用来引导,通常基因组点突变只需要改变一个碱基,因此修复模板与基因组上待编辑片段序列几乎没有差别,因此会被sgrna靶向而被cas9蛋白切割而影响点突变编辑效率。现有的编辑系统,如dcas9蛋白,其点突变存在局限性。本发明将sgrna上的20bp的crrna进行同义突变,将避免sgrna的靶向以此防止cas9蛋白的切割来提高点突变编辑效率。
29.2)利用上述编辑系统对枯草芽孢杆菌基因组seca的e646位点进行了理性改造,此部分研究操作简单,没有复杂的分子操作以及大量的工作,并提高了枯草芽孢杆菌对碱性蛋白酶的分泌能力。相比于通过信号肽筛选以及修饰来提高异源蛋白的分泌效率,本发明工作量小,方向性强,是一种全新的方法,为以后人们的改造枯草分泌系统提供了新策略。
30.3)本发明基于最简单的crispr/cas9系统进行高效的枯草芽孢杆菌基因组点突变,通过改造枯草芽孢杆菌sec途径的关键组分seca提高对异源蛋白的分泌能力,为以后人们进行基因组点突变提供新的方法。
附图说明
31.图1示出了seca与secy复合结构示意图;
32.图2示出了酪氨酸标准曲线;
33.图3示出了pjoe8999质粒图谱;
34.图4示出了枯草芽孢杆菌同源重组示意图;
35.图5示出了pjoe8999-secagrna质粒示意图;
36.图6示出了pjoe8999-t20secagrna质粒示意图;
37.图7示出了测序比对结果;
38.图8示出了基因组seca基因crrna编辑模板上下游同源臂片段及融合片段的核酸电泳图;其中,a:上下游片段体外扩增结果,泳道2:上游片段1500bp,泳道3:下游片段1000bp;b:融合pcr结果,泳道2:融合片段;
39.图9示出了seca基因crrna编辑结果;其中,a:pcr扩增seca基因琼脂糖凝胶电泳验证;b:测序结果比对;
40.图10示出了t20-crrna点突变编辑效率;
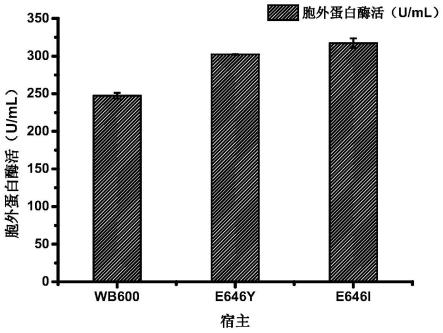
1.图11示出了突变体摇瓶发酵及胞外酶活检测结果。
具体实施方式
2.以下结合具体实施例,对本发明做进一步说明。应理解,以下实施例仅用于说明本发明而非用于限制本发明的范围。
3.1材料与方法
4.1.1菌株与质粒
5.本发明所用的质粒与菌株见表1。
6.表1质粒与菌株质粒与菌株特点与用途pma5-sd-9nt-apre蛋白质表达质粒pjoe8999crispr/cas9质粒dh5α用于基因克隆wb600蛋白表达宿主wb600 t20用于枯草杆菌基因组编辑e646y,e646i经过sec途径改造的宿主
7.1.2主要试剂配制
8.氨苄青霉素(amp):使用电子分析天平称取氨苄青霉素粉末1.000g,并将其溶于去离子水中,待粉末完全溶解之后,将溶液转移至10.00ml容量瓶中定容,最终配制为100mg/ml的amp溶液,使用孔径为0.45μm的无菌滤膜过滤除菌,分装无菌ep管中,放置在-20℃保存。
9.卡那霉素(kan):使用电子分析天平称取卡那霉素粉末0.500g,并将其溶于去离子水中,待粉末完全溶解之后,将溶液转移至10.00ml容量瓶中定容,最终配制为50mg/ml的kan溶液,使用孔径为0.45μm的无菌滤膜过滤除菌,分装无菌ep管中,放置在-20℃保存。
10.1.3主要试剂
11.本实验主要使用的试剂见表2。
12.表2实验所用的主要试剂试剂限制性内切酶
t4 dna ligasertaq2xprimestar maxkod onehieff clone
tm one step cloning kitdna marker质粒提取试剂盒胶回收试剂盒casein福林酚(br)三氯乙酸dna纯化试剂盒氨苄青霉素卡那霉素
13.1.4主要培养基配方
14.lb液体培养基:氯化钠:10g/l;酵母提取物:5g/l;蛋白胨:10g/l。
15.lb固体培养基:在lb液体培养基中加入1.5%-2%的琼脂粉。
16.1.5引物序列
17.实验中所用到的引物见表3。
18.表3引物序列表
19.1.6标准曲线的绘制
20.以去离子水为溶剂,分别吸取100μg/ml l-酪氨酸标准溶液0ml,1ml,2ml,3ml,4ml,5ml,配置成浓度为0μg/ml,10μg/ml,20μg/ml,30μg/ml,40μg/ml,50μg/ml的酪蛋白标准溶液。
21.分别吸取上述溶液各1.00ml(需做三组平行实验)于玻璃试管中,接着向每根试管中加入5.00ml 0.4m碳酸钠溶液和1.00ml福林试剂使用液,混合均匀后将试管放置于40℃水浴锅中显色20min,显色结束取出,使用分光光度计于680nm下测吸光值a,以吸光值a为纵坐标,酪氨酸浓度c为横坐标,绘制标准曲线(此线应通过零点),如图2所示。
22.2实验方法
23.2.1常规分子生物学实验操作
24.2.1.1常规pcr
25.参考rtaq,prime star max,kod one等说明书,进行pcr扩增程序的设定。
26.rtaq/prime star max pcr体系如下表4所示。
27.表4
28.kod one pcr体系如下表5所示。
29.表5基因模板(菌液)1μl2x prime kod one25μl引物11.5μl引物21.5μl加ddh20至50μl
30.大肠杆菌的菌落pcr验证使用rtaq酶。
31.2.1.2融合pcr
32.ab两片段的融合pcr,a片段的3’端与b片段的5’端的25个碱基序列相同,反应体系如下表6所示。
33.表6
34.参考说明书进行pcr,使用胶回收试剂盒进行胶回收,分别获得a,b片段,回收产物于-20℃保存备用。反应体系如下表7所示。
35.表7pcr—ab片段 模板a与b加入相同摩尔量模板b与a加入相同摩尔量a正向引物1μlb反向引物1μl2x prime star max25μl加ddh2o至50μl
36.参考说明书进行pcr,获得ab片段融合之后的pcr产物,借助胶回收试剂盒进行胶回收,回收产物于-20℃保存。
37.2.1.3 dna电泳胶
38.1.2%琼脂糖胶:称取1.5g琼脂糖放置于250ml锥形瓶中,量取120ml 1xtae溶液加入锥形瓶中,使用微波炉加热至琼脂糖完全溶解,使用自来水稍稍冷却后,加入核酸染料,摇匀后,倒入到插好梳子的制胶模具中,待胶完全冷却凝固后,使用刀具切好放置于4℃冰箱保存。
39.核酸电泳:将核酸胶放置于展开的pe手套上,使用移液枪吸取1 x tae电泳缓冲液将核酸胶上的上样孔注满,注意不留气泡,接着在pe手套上点加5 x loading buffer,每个点约1.5μl的5 x loading buffer,移液枪吸取2-3μl dna样品与上样缓冲液混合均匀,然后将所有混合液加入到核酸胶上样孔中,接着在电泳槽中加入适量的1 x tae电泳缓冲液,然后将胶置于其中,在120v条件下电泳20min,紫外分析仪下拍照观察结果。
40.2.1.4 dna片段的胶回收
41.参考hipure gel pure dna kits说明书。
42.2.1.5 dna片段的纯化
43.参考hipure pcr pure kits说明书。
44.2.1.6质粒dna的抽提
45.参考axyprep质粒dna小量试剂盒。
46.2.1.7酶切实验
47.质粒酶切体系如下表8所示。
48.表810xfd buffer4μl待切质粒3μgfd限制性内切酶12μlfd限制性内切酶22μl加ddh2o至40μl
49.37℃酶切1.5~2h。
50.dna片段酶切体系如下表8所示。
51.表910xfd buffer3μl待切dna片段3μgfd限制性内切酶11.5μlfd限制性内切酶21.5μl加ddh2o至30μl
52.37℃酶切2h,胶回收需要的片段。
53.2.1.8酶切质粒与dna片段的连接
54.连接体系(10μl)如下表10所示。
55.表1010xligase buffer1μlt4 dna ligase1μldna片段质粒载体摩尔数的2-3倍质粒载体100-200ng
加ddh2o至10μl
56.22℃连接2h或16℃连接过夜,转入dh5α感受态细胞进行质粒的扩增。
57.2.1.9无缝克隆
58.体系配制如下表11所示。
59.表11
60.转入dh5α感受态细胞进行质粒的扩增。
61.2.2枯草芽孢杆菌wb600宿主转化试剂配制
62.(1)spⅰsalts solution
63.使用电子分析天平分别称取硫酸铵2g,三水合磷酸氢二钾14g,磷酸二氢钾6g,柠檬酸三钠二水合物1g置于烧杯中,加入去离子水溶解,待其完全溶解后转移至500ml容量瓶中定容,121℃灭菌20min,室温保存。
64.(2)spⅱsalts solution
65.使用电子分析天平分别称取七水合硫酸镁0.2g置于烧杯中,加入去离子水溶解,待其完全溶解后转移至500ml容量瓶中定容,121℃灭菌20min,室温保存。
66.(3)100x caye培养基
67.使用电子分析天平分别称取酪蛋白水解物2g,酵母提取物10g置于烧杯中,加入去离子水溶解,待其完全溶解后定容至100ml,121℃灭菌20min,4℃保存。
68.(4)50mm cacl2溶液,250mm mgcl2溶液,10mm egta,50%葡萄糖
69.使用电子分析天平称取0.555g cacl2颗粒溶于去离子水中,待其完全溶解之后定容至100ml。
70.使用电子分析天平称取2.3804g mgcl2粉末溶于去离子水中,待其完全溶解之后定容至100ml。
71.使用电子分析天平称取0.4g egta粉末溶于去离子水中,待其完全溶解之后定容至100ml,溶解时需加入少量氢氧化钠使ph=8.0。
72.称取50g无水葡萄糖,加热溶解于少量去离子水中,待其冷却至室温定容至100ml。
73.注:除了葡萄糖需115℃,20min灭菌外,其余试剂均需121℃,20min灭菌。
74.(5)mediumⅰ(20ml)
75.分别吸取9.8ml spⅰsalts solution,9.8ml spⅱsalts solution,200μl 50%葡萄糖和200μl 100x caye溶液于无菌试管中混合均匀,备用。此培养基需现制现用。
76.(6)mediumⅱ(6ml)
77.分别吸取5.88ml mediumⅰ,60μl 50mm cacl2溶液和60μl 250mm mgcl2溶液于另一无菌试管中,混合均匀,备用。此培养基需现制现用。
78.2.3枯草芽孢杆菌wb600宿主转化
79.(1)前一天晚上在lb平板上挑选wb600宿主单克隆(由本实验室保存),接种于5ml液体lb培养基试管中,在37℃,220rpm摇床中培养12-14小时;
80.(2)吸取200μll过夜培养的新鲜菌液接种于含5ml sp i培养基的试管中,在37℃,220rpm摇床中培养,培养4-4.5h后测菌体od600nm,当od600nm达到1.5时停止培养;
81.(3)快速吸取200μl新鲜spⅰ培养液接种于含有3ml spⅱ培养基的试管中,在37℃,220rpm摇床中培养,培养1-1.5小时后,取样测od600nm,当od600nm达到0.50-0.55时,停止培养;
82.(4)按1%(v/v)的量向试管中加入100x egta溶液,将其置于37℃,220rpm摇床继续培养,培养10min(准确计时)后取出,便可获得枯草芽孢杆菌wb600感受态;
83.(5)将感受态细胞分装于1.5ml无菌离心管中,每管分装800μl,同时向管中加入500ng质粒,轻轻拍打混匀,于37℃,220rpm摇床中培养2.5小时;
84.(6)培养结束后,以4500rpm离心4min收集菌体,用移液枪吸取700μl上清液丢掉,留100μl重悬菌体,涂布在含有相应抗生素的lb固体培养基平板上,37℃过夜培养。
85.注:感受态现制现用,不可甘油保存。
86.2.4蛋白酶活性测定
87.2.4.1配制主要试剂
88.(1)0.4m碳酸钠溶液
89.称取无水碳酸钠(na2co3)粉末42.4g,溶于去离子水中,溶解后定容至1000ml
90.(2)0.4m三氯乙酸溶液。
91.称取三氯乙酸(ccl3cooh)65.4g,溶于去离子水中,溶解后定容至1000ml。
92.(3)0.5m氢氧化钠溶液,1m盐酸溶液和0.1m盐酸溶液
93.依据gb601进行配制。
94.(4)福林试剂使用液
95.将福林酚试剂按照福林酚试剂:去离子水=1:2的比例稀释,即福林试剂使用液。
96.(5)硼酸缓冲溶液(ph 10.5)
97.a液:称取硼酸钠19.08g溶于去离子水中,溶解后定容至1000ml;
98.b液:称取氢氧化钠4.0g溶于去离子水中,溶解后定容至1000ml;
99.量取a液500ml,b液400ml置于烧杯中搅拌混匀,加去离子水稀释至1000ml,最后使用ph计调节ph至10.5。
100.(6)10g/l酪蛋白溶液
101.使用电子分析天平准确称取1.000g酪蛋白粉末,先用少量0.5m氢氧化钠溶液浸湿粉末,随后加入约70ml硼酸缓冲液溶解酪蛋白,需在沸水浴中边加热边搅拌,直至溶液中无细小酪蛋白颗粒为止,将其放置室温冷却,冷却至室温后将其转移至100ml容量瓶中用硼酸缓冲液定容。将其转移至蓝盖瓶中置于4℃冰箱保存,有效期3天。
102.(7)100μg/mll-酪氨酸标准溶液
103.首先使用电子分析天平准确称取预先干燥至恒重的l-酪氨酸0.1000g,将其溶解于1m盐酸溶液中并定容至100ml,即为1mg/ml酪蛋白标准溶液;然后吸取10ml酪蛋白标准溶液于容量瓶中,用0.1m盐酸溶液稀释至刻度,即得到100μg/ml l-酪蛋白标准溶液。
104.2.4.2蛋白酶活性测定
105.(1)将10g/l酪蛋白溶液放置于40℃恒温水浴锅中预热;
106.(2)将发酵液在4℃条件下,12000rpm离心10min,收取上清液,此为稀释前粗酶液;
107.(3)使用硼酸缓冲液将粗酶液稀释10-20倍,待测酶活性;
108.(4)分别吸取1ml稀释后粗酶液于4支玻璃试管中(其中1支为空白样,3支为平行实验组),置于40℃水浴锅中预热2min;
109.(5)向空白对照样试管中加入2.00ml三氯乙酸溶液,实验组试管中分别加入1ml酪蛋白溶液,摇匀,40℃水浴10min;
110.(6)水浴结束后,快速向实验组试管中加入2ml三氯乙酸溶液,并摇匀,空白对照组中加入1ml酪蛋白溶液并摇匀;
111.(7)从4支试管中分别吸取2ml混合液于2ml ep管中,12000rpm离心5min;
112.(8)分别取出1ml上清液于空试管中,加入5ml碳酸钠溶液,混合均匀;
113.(9)继续向试管中加入1ml福林试剂使用液,在40℃水浴锅中显色20min;
114.(10)显色结束后,于680nm波长,用10mm比色皿测其吸光度a,依据标准曲线计算酶活性。
115.2.5 crispr/cas9系统
116.质粒pjoe8999是一种大肠-枯草穿梭载体,pjoe8999质粒图谱如图3所示,含有在大肠杆菌中的最小复制起点pucori和在枯草芽孢杆菌中温度敏感型复制起点pe194ts。其所携带的卡那霉素抗性基因能够在这两种细菌中起作用。对于基因组编辑,质粒pjoe8999携带的是在甘露糖启动子pmanp下游能被甘露糖诱导的cas 9基因。并且质粒含有通过强启动子转录的单个引导rna(sgrna)编码序列。这种sgrna将引导cas9核酸酶到达其靶点。位于sgrna序列5’末端的20bp靶序列位于bsai位点之间,负责靶标特异性。donor dna可被插入至两个sfiⅰ位点之间,用于基因组修复。
117.2.6枯草芽孢杆菌基因组编辑
118.将构建完成的crispr/cas9质粒转化枯草芽孢杆菌宿主中,将其涂布在含有5μg/ml卡那霉素和0.2%甘露糖的lb琼脂平板上,于30℃恒温培养箱中培养48h,待其培养24h后,取出平板,在平板上标记出已生长出来的菌落,放回培养箱继续培养48h(若培养24h未长出任何菌落,可选取48h内生长出的菌落)。培养结束,将选出的菌落使用枪头点样在无抗琼脂平板上,在50℃条件下培养16-18h,丢去质粒。最后将菌落按照对应的位置点样在含有50μg/ml卡那霉素琼脂平板上过夜培养,进行丢质粒验证。
119.实施例1改造crispr/cas9系统,构建t20宿主获得优化后的crispr/cas9
‑‑
t20系统
120.3.1 crispr/cas9系统的操作原理及过程
121.3.1.1 crrna序列的获得
122.本实验需借助https://cctop.cos.uni-heidelberg.de/网站设计靶向枯草芽孢杆菌基因组的crrna。在进行crrna设计之前,我们需要从ncbi网站获取seca基因序列。然后将需要靶向的seca序列复制在query sequence框中,同时在species框中选中bacillus subtilis subsp.subtilis str.168,最后单击submit按钮进行设计。然后会获得一系列网站所建议的crrna序列。网站会评价这些序列并标记分数,可选择高分数序列作为crrna。
123.3.1.2 sgrna表达质粒的构建
124.grna构建过程中,需要将crrna插入至tracrrna上游,即启动子与tracrrna之间,由crrna与tracrrna共同组成sgrna。crrna是长度为20bp的靶向序列,只需在设计引物时,直接将crrna序列设计在前后引物的5’端。使用反向引物secagrna-r从启动子的3’方向开始反向扩增,用正向引物secagrna-f从tracrrna的5’正向扩增,然后以含pjoe8999的dh5α新鲜菌液为模板,使用kod one dna聚合酶进行pcr扩增,最后按照构建pma5-sd-9nt-293sp-apre质粒的方法
125.使用引物secagrna-f,secagrna-r,以pjoe8999质粒(本实验室保存)为模板,使用kod one dna聚合酶进行pcr,对pcr产物进行胶回收,回收产物使用无缝克隆酶连接成环状质粒,则pjoe8999-secagrna构建完成。即对纯化后的pcr产物进行无缝克隆来构建sgrna表达质粒。
126.3.1.3重组交换整入目的基因
127.crispr/cas9质粒上不仅需要sgrna表达框,还需要donor dna来修复被cas9切割的基因组。将crispr/cas9质粒导入至枯草杆菌细胞内后,sgrna可以与cas9蛋白结合,随后对含有pam序列的靶dna序列进行切割,随后依赖枯草芽孢杆菌的同源重组完成目的基因的整入,这个过程需要两侧同源臂的参与。
128.枯草芽孢杆菌同源重组示意图如图4所示。cas9蛋白在sgrna的引导下,会在靶dna位点上停留并与之结合,随后对基因组dna进行切割,造成dna双链的断裂。同源重组是枯草杆菌对基因组dna损伤主要的修复方式。此时需要存在于donor dna两端的上下游同源臂与dna双链断裂处进行同源重组。此上下游同源臂分别长为500-1000bp。因此,在枯草杆菌基因组编辑过程中,dna被切割后,借助修复系统以上游同源臂-目的dna序列-下游同源臂为模板对基因组进行修复,以此完成靶基因的删除及目的基因的整合。
129.3.2 sgrna靶向质粒的构建
130.采用3.1.1中提到的方法,首先,从ncbi网站中查询seca基因序列,其基因序列在ncbi中的reference sequence为nc_000964.3。然后,将seca基因输入crrna设计网站中,以此获得网站推荐的crrna序列。根据网站所标记的分数,选取的分数最高的crrna序列。
131.pjoe8999是已经商品化的crispr/cas9质粒,本课题所用质粒由鲁华所王学东老师赠与。该质粒含有能够在大肠杆菌与枯草杆菌中起作用的卡那霉素抗性基因筛选标记。cas9基因可被甘露糖进行诱导表达。并且质粒含有强启动子转录的sgrna表达盒。将crrna序列插入至tracrrna的5’端,以含pjoe8999的dh5α新鲜菌液为模板,用secagrna-f引物和secagrna-r引物进行pcr扩增。
132.pcr体系如下:
133.将pcr产物进行琼脂糖凝胶电泳验证,条带大小符合,进行胶回收,并将回收的dna
片段命名为pjoe8999-secagrna。对此片段自身按照以下体系和条件进行无缝克隆连接实验。
134.连接体系如下:2x in-fusion hd enzyme premix5μl线性化pjoe8999-secagrna质粒150ng加ddh2o至10μl置于50℃反应30min。 135.常规的dh5a转化方法:
136.从-80℃冰箱中快速取出dh5α感受态细胞并将其放置于冰上,待感受态细胞融化后快速加入待转化的连接体系或者200ng质粒,此过程不宜超过5min。注意禁止用无菌枪头吹打混匀,可用手指轻轻弹撞ep管混匀,随后冰浴15-20min。
137.(2)冰浴结束,将感受态细胞快速放置于42℃水浴锅中热激90s,然后快速将其转移到冰上,冰浴5min,此过程尽量不要摇晃感受态细胞,防止转化效率降低。
138.(3)随后加入600μl无菌lb培养基(不含抗生素),置于37℃、220rpm摇床中培养60min。
139.(4)培养结束,室温条件下5000rpm离心4min,使用移液枪吸出600μl上清丢弃,然后使用无菌枪头将菌体沉淀重悬,将100μl菌液全部涂布于相应抗生素的lb平板上,放置于37℃培养箱中过夜培养,待其长出大肠杆菌菌落。
140.将无缝克隆连接体系按照常规的dh5a克隆宿主转化方法的方法转化至克隆宿主dh5α中,并且涂布于含有50μg/ml的卡那抗生素抗性的lb平板上,第二天挑选阳性克隆培养后送去测序,把测序结果通过snapgene软件与绘制的质粒图谱进行比对分析,将测序结果正确的质粒命名为pjoe8999-secagrna。pjoe8999-secagrna质粒示意图如图5所示。
141.3.3 crrna序列同义突变设计
142.由于密码子具有简并性,因此可以对crrna进行同义突变,改变核酸序列,但是不变氨基酸序列,不影响seca的功能。因此,在构建用于同源重组的编辑模板时,将原有crrna序列同义突变,突变后的crrna序列称为t20-crrna。将原crrna序列尽最大可能变为与原序列不同的t20-crrna序列,以防止sgrna的靶向识别。将突变后crrna序列通过设计引物将突变后的序列引入。
143.3.4 donor dna贡献质粒的构建
144.分别设计两对引物,对seca上下游同源臂进行扩增。首先设计上游同源臂扩增引物,其正向引物同时引入sfiⅰ限制性酶切位点,反向引物则需含有t20-crrna序列;同理,下游同源臂的正向引物含有t20-crrna序列,反向引物引入sfiⅰ酶切位点。
145.使用引物seca-sfi
ⅰ‑
f与t20-r对seca上游同源臂进行pcr,引物seca-sfi
ⅰ‑
r与t20-f对下游同源臂进行pcr。所得到的pcr产物在进行琼脂糖凝胶电泳验证正确后,进行胶回收,获得纯净的上、下游同源臂dna片段。
146.以上、下游同源臂为模板,seca-sfi
ⅰ‑
f/seca-sfi
ⅰ‑
r为引物,使用kod one进行融合pcr,体外扩增程序按照kod one说明书进行设定。融合pcr体系如下表12所示。
147.表12上游同源臂50ng
下游同源臂50ngseca-sfi
ⅰ‑
f1.5μlseca-sfi
ⅰ‑
r1.5μl2x kod one25μlddh2oadd to 50μl
148.融合pcr结束后,进行琼脂糖凝胶电泳验证,条带符合上下游加一起大小即认为融合成功,之后进行胶回收,获得纯净的融合后dna片段。
149.将含有pjoe8999-secagrna质粒的大肠杆菌克隆宿主甘油菌接种于含有5ml lb培养基的试管中,加入5μl的卡那抗生素,培养12-14h后,按照2.2.1.6方法抽提质粒,使用nano测其质粒浓度,置于4℃保存备用。
150.对融合片段和pjoe8999-secagrna质粒进行sfiⅰ酶切,使其具有粘性末端,随后使用t4连接酶进行连接。
151.融合片段酶切体系如下表13所示。
152.表1310xfd buffer1μl待切融合片段1-1.5μgsfiⅰ1μl加ddh2o至10μl
153.50℃酶切2h,胶回收需要的片段。同时酶切3个体系,以此获得充足的酶切融合片段。
154.质粒酶切体系如下表14所示。
155.表14
156.50℃酶切2h,胶回收需要的片段。同时酶切3个体系,以此获得充足的酶切质粒骨架。
157.将酶切融合片段体系和酶切质粒体系分别进行琼脂糖凝胶电泳,将所需条带切下,并按照2.2.1.4方法胶回收,获得具有粘性末端的融合片段和质粒,置于-20℃保存备用。
158.使用t4连接酶将融合片段与pjoe8999-secagrna质粒骨架进行连接,连接体系如下表15所示。
159.表1510x ligase buffer1μlt4 dna ligase1μl融合片段120ng
pjoe8999-secagrna质粒载体140ng加ddh2o至10μl
160.22℃连接2h。
161.常规的dh5a转化方法:
162.从-80℃冰箱中快速取出dh5α感受态细胞并将其放置于冰上,待感受态细胞融化后快速加入待转化的连接体系或者200ng质粒,此过程不宜超过5min。注意禁止用无菌枪头吹打混匀,可用手指轻轻弹撞ep管混匀,随后冰浴15-20min。
163.(2)冰浴结束,将感受态细胞快速放置于42℃水浴锅中热激90s,然后快速将其转移到冰上,冰浴5min,此过程尽量不要摇晃感受态细胞,防止转化效率降低。
164.(3)随后加入600μl无菌lb培养基(不含抗生素),置于37℃、220rpm摇床中培养60min。
165.(4)培养结束,室温条件下5000rpm离心4min,使用移液枪吸出600μl上清丢弃,然后使用无菌枪头将菌体沉淀重悬,将100μl菌液全部涂布于相应抗生素的lb平板上,放置于37℃培养箱中过夜培养,待其长出大肠杆菌菌落。
166.将t4连接体系按照常规的dh5a转化方法的方法转化至克隆宿主dh5α中,并且涂布于含有50μg/ml的卡那抗生素抗性的lb平板上,置于37℃培养箱中过夜培养,第二天从平板上筛选阳性克隆,使用seca-sfi
ⅰ‑
f/seca-sfi
ⅰ‑
r引物进行菌落pcr验证,验证成功的菌落送去测序,测序结果使用snapgene软件与目的片段进行比对,比对结果正确的菌株命名为pjoe8999-t20secagrna。pjoe8999-t20secagrna质粒示意图如图6所示。
167.按照pjoe8999-secagrna质粒的抽提方法,提取pjoe8999-t20secagrna质粒,置于-20℃冰箱中备用。
168.3.5 wb600基因组编辑
169.按照2.2.3中wb600感受态制备方法制备wb600感受态细胞,将构建完成的pjoe8999-t20secagrna质粒转化枯草杆菌感受态细胞中,将其涂布在含有5μg/ml卡那霉素和0.2%甘露糖的lb平板上,先于30℃恒温培养箱中培养24h,然后取出平板,在平板上标记出已生长出来的菌落,之后放回培养箱继续培养48h。培养结束后,将标记的菌落点样在不含抗生素的lb平板上,在50℃条件下培养16-18h,丢去质粒。最后将菌落对应点样于含有50μg/ml卡那霉素lb平板上过夜培养,验证质粒是否丢失。
170.3.6编辑结果验证
171.验证基因组t20-crrna宿主。在确认基因组编辑完成,宿主质粒完全丢失后,以seca-sfi
ⅰ‑
f/seca-sfi
ⅰ‑
r为引物,扩增编辑后宿主的seca基因,并进行琼脂糖凝胶电泳验证pcr结果,获得目的条带后将pcr产物送去测序,测序结果用snapgene软件比对,结果正确则将此宿主命名为t20。
172.3.7 t20-crrna编辑效率验证
173.若t20宿主构建成功,需对t20-crrna序列进行编辑效率验证。将t20-crrna序列插入至tracrrna,将质粒命名为pjoe8999-t20secagrna,接着将野生型seca基因作为同源模板用于修复双链的断裂。对t20宿主的seca基因进行编辑,编辑后宿主在丢质粒后随机选取多个菌落进行seca基因扩增,并送去测序,比对测序结果,记录编辑成功菌落个数,以此计算编辑成功率。
174.4.1实验结果与讨论
175.4.1.1 sgrna靶向质粒的构建
176.将seca基因输入crrna设计网站后,获得了一系列网站推荐的crrna序列。根据网站所标记的分数,选取的分数最高的crrna序列:gaaggtgtaaaagagcttgg。将此序列通过引物插入tracrrna的5’端,测序结果表明crrna序列成功插入,见图7,pjoe8999-secagrna质粒构建成功。
177.应当理解的是,本实施例方法只是提供了评分最高的crrna序列,其余的crrna序列均可。
178.4.1.2 crrna同义突变设计
179.基于密码子具有简并性,本实施例对crrna序列进行了同义突变,改变了crrna核酸序列,即将gaaggtgtaaaagagcttgg(seq id no.13)修改为gagggggtcaaggaattggg(seq id no.14),将修改后的序列通过基因组编辑,完成seca基因crrna序列的替换。
180.4.1.3 donor dna质粒的构建
181.4.1.3.1修饰基因组seca crrna序列的编辑模板构建
182.上、下游同源臂扩增成功,并且成功将上、下游同源臂通过融合pcr连接成一条dna分子,见图8。
183.4.1.4 wb600基因组编辑结果
184.4.1.4.1 seca基因crrna同义突变结果
185.在基因编辑完成和确认质粒丢除完全后,挑选菌落进行pcr扩增seca基因通过琼脂糖凝胶电泳验证后(图9中的a所示),将pcr产物送去测序,测序结果显示,t20宿主构建成功(图9中的b所示),可以从比对结果中看出,核酸序列发生显著变化,而氨基酸序列并没有改变。通过编辑效率验证,如图10所示,t20-crrna编辑效率可到达86%,即挑选7个成功丢质粒的菌落中,有6个菌落编辑成功。其编辑效率可满足要求,因此,宿主t20可作为接下来基因组编辑研究宿主。
186.实施例2借助改造的crispr/cas9系统对t20宿主进行基因点突变获得改造宿主e646y、e646i
187.seca上的e646会与疏水性的磷脂进行相互作用。疏水性最强带有脂肪族侧链的异亮氨酸i,和亲水性带有芳香族侧链的酪氨酸y对带有较弱负电性的谷氨酸e进行替换,旨在使seca与膜蛋白(膜蛋白形成了膜上的分泌通道)结合更稳定,以提高蛋白分泌能力。
188.5.1同源模板点突变引入
189.首先通过pcr对突变位点上游和下游片段进行扩增。使用引物seca-sfi
ⅰ‑
f/e646-r扩增突变位点e646上游片段,命名为e646-up,使用引物seca-sfi
ⅰ‑
r/e646-f扩增突变位点e646下游片段,命名为e646-down。
190.接着通过设计引物引入突变,使用2对引物seca-sfi
ⅰ‑
f/e646y-r,seca-sfi
ⅰ‑
f/e646i-r,以e646-up为模板,获得已经引入突变的2条dna片段,分别命名为e646y-up,e646i-up。
191.最后通过融合pcr对上下游片段进行融合,获得同源模板。以融合e646y同源模板为例。按照以下pcr体系加入模板,引物和dna聚合酶。
192.pcr体系如下表16所示。
193.表16e646y-up70nge646-down180ngseca-sfi
ⅰ‑
f1μlseca-sfi
ⅰ‑
r1μl2x prime star max25μl加ddh2o至50μl
194.参考prime star max说明书进行pcr程序设定。
195.融合结束后,将融合产物进行琼脂糖凝胶电泳验证,验证正确后按照2.2.1.4的方法进行胶回收,获得大小约为2500bp的突变后seca序列e646y,并将以其为编辑模板进行基因组断裂的修复同时引入突变点。seca突变体e646i按同样的方式进行融合pcr最终获得同源模板。
196.5.2 crispr/cas9质粒的构建
197.将含有3.7中所构建的pjoe8999-t20secagrna质粒的dh5α接种于含有5ml lb培养基的试管中,过夜培养,按照2.1.6中质粒提取方法,抽提质粒。
198.按照2.1.7中的酶切体系和方法,使用sfiⅰ酶切质粒和2种同源模板(获得大小约为2500bp的突变后seca序列e646y,并将以其为编辑模板进行基因组断裂的修复同时引入突变点。seca突变体e646i按同样的方式进行融合pcr最终获得同源模板),酶切之后进行琼脂糖凝胶电泳,除去杂质和多余的dna小片段,得到含有碱基互补粘性末端的dna片段和线性pjoe8999-t20secagrna质粒。
199.按照2.1.8中的t4连接酶连接的方法,将dna片段与线性pjoe8999-t20secagrna质粒连接,获得crispr/cas9基因组编辑质粒,并分别命名为pjoe8999-t20secagrna-e646y,pjoe8999-t20secagrna-e646i。
200.5.3宿主基因组编辑及突变体筛选
201.按照3.5中wb600宿主基因组编辑方法对t20宿主基因组进行编辑。
202.基因组编辑结束后,使用引物seca-sfi
ⅰ‑
f/seca-sfi
ⅰ‑
r分别扩增2组基因组编辑后菌落的seca基因。经过琼脂糖凝胶电泳验证后,将pcr产物送去测序。使用snapgene软件将测序结果与目的序列进行比对,基因组编辑正确的突变宿主进行甘油保存,并分别命名为e646y和e646i。
203.5.4突变体摇瓶发酵及胞外酶活检测
204.将pma5-sd-9nt-293sp-apre质粒(本实验室保存)按照2.2.3枯草芽孢杆菌wb600宿主转化中枯草杆菌转化方法分别转化至e646y和e646i突变宿主感受态细胞中,并分别将其涂布于牛奶平板中,置于37℃培养箱中过夜培养,第二天挑选出现透明圈的菌落于含有5ml lb培养基的试管中,并加入5μl卡那抗生素。培养约10-12h后,进行甘油保存,并测od600nm。将剩余新鲜菌液按照最终od600nm为0.02接种于含有100ml lb的250-ml摇瓶中,并加入100μl卡那抗生素进行发酵培养。
205.5.5枯草芽孢杆菌发酵条件
206.先把重组菌的甘油菌在含有卡那抗生素的固体培养基上划线活化,然后挑选单菌落于含有5ml培养基的试管中,37℃条件下培养10-12h。
207.将试管中的新鲜菌液稀释10倍,测od600nm。
208.按照初始od600nm为0.021转接至含有100ml lb培养基的250ml摇瓶中进行培养,发酵条件为37℃,220rpm。
209.每隔6h取样一次,测其od600nm及胞外粗酶活。
210.5.6胞外粗酶活检测
211.按照2.4中的蛋白酶活性测定方法测定发酵液中蛋白酶酶活。
212.按照5.5中重组菌发酵条件与2.4中胞外酶活检测方法对以上2种突变体重组菌e646y和e646i分别进行酶活检测。
213.5.7结果
214.突变体摇瓶发酵及胞外酶活检测
215.如图11所示,在整个发酵周期中,突变体e646i的酶活提高较高,在酶活最高点相对于野生型提高了28.2%;e646y提高了22.2%。
216.以上所述的,仅为本发明的较佳实施例,并非用以限定本发明的范围,本发明的上述实施例还可以做出各种变化。凡是依据本发明申请的权利要求书及说明书内容所作的简单、等效变化与修饰,皆落入本发明专利的权利要求保护范围。本发明未详尽描述的均为常规技术内容。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
