1.本发明属于医药化工领域,具体涉及一种替莫唑胺的制备方法。
背景技术:
2.替莫唑胺(temozolomide),化学名为8-氨基甲酰基-3-甲基咪唑[5,1-d]-1,2,3,5-四嗪-4(3h)-酮,分子式:c6h6n6o2;分子量:194.15;cas登记号:85622-93-1,结构式如下:
[0003][0004]
替莫唑胺最早由英国阿斯顿大学公司开发,后由德国先灵葆雅制药获开发权,于1999年在美国上市。药理学研究证明,替莫唑胺为是一种对脑胶质瘤有较好疗效的新型药物;具有生物利用度高,可口服,易于透过血脑屏障,与其他药物比没有叠加毒性,具有较宽的抗肿瘤谱。目前,替莫唑胺是较好的治疗脑胶质瘤和恶性黑色素瘤的抗癌药物,替莫唑胺的胶囊剂已在欧美批准用于治疗恶性神经胶质瘤。
[0005]
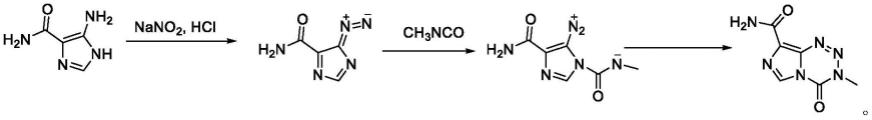
替莫唑胺的常规合成方法是以5-氨基-1h-咪唑-4-甲酰胺或其盐酸盐为原料与亚硝酸钠反应,经重氮化再与异氰酸甲酯进行反应制得替莫唑胺(journal of the chemical society,perkin transactions 1,1998,10:l669-1775)其合成路线如下。此方法原子利用率较高,因而得到高纯度和高收率的产物替莫唑胺,但合成过程中试用了毒性较大的异氰酸甲酯,操作风险较大,不易生产合成。
[0006][0007]
第二类方法为异氰酸甲酯替代法:一种是用3-羧基替莫唑胺去质子化法合成替莫唑胺,收率为26%(journal of the chemical society,chemistry commum,19914,14:1687-1688);另一种是用3-三甲基硅烷甲基咪唑四嗪与氟化四丁基胺在乙腈-乙酸混合溶剂中反应得到替莫唑胺,收率78%(biochemistry,1994,33(31):9045-9051)。这两种方法虽然避免了直接使用异氰酸甲酯,但因替代物不太稳定,收率均不高。
[0008][0009]
第三类方法为甲氨基甲酰氯的合成方法(journal of the organic chemistry,1997,62:7288-7294)。从5-氨基咪唑-4-甲酰胺出发,合成了5-氨基-1-(n-甲基氨基甲酰
基)咪唑-4-甲酰胺,再通过重氮化反应得到替莫唑胺。该方法使用中等毒性甲胺基甲酰氯替代剧毒的异氰酸甲酯,且5-氨基-1-(n-甲基氨基甲酰基)咪唑-4-甲酰胺具有多个重氮化反应位点,该步骤将产生异构体副产物,反应收率有待提高。
[0010][0011]
第四类方法为不使用5-重氮基咪唑-4-甲酰胺和异氰酸甲酯的新方法(journal of the chemical society,perkin transactions 1,2002,16:1877-1880),5-硝基咪唑在硝酸存在的条件下与氰酸钾反应生成5-硝基咪唑4-乙腈,然后经过多步反应生成替莫唑胺。该法收率较高,但合成路线较长,合成路线太过复杂,不适合工业生产。
[0012][0013]
第五类方法为羰基二咪唑参与反应的新方法(wo2018/112589):以羰基二咪唑出发经历两次咪唑环交换,再重氮化关环得到替莫唑胺。该方法试剂毒性低,易操作,但中间体n-甲基-1-h-咪唑甲酰胺须硅胶柱纯化,操作复杂。同时,需重氮化的中间体化合物具有双反应位点,关环得到两个产物,使得此步收率降低。
[0014][0015]
综上所述,现有替莫唑胺制备方法存在合成路线长、收率低、纯度低以及技术要求高、环境污染严重、中间体不稳定、生产成本高的问题。因此,为替莫唑胺探究一条操作简便、收率更高、更适合工业化生产的工艺路线仍然是目前需要解决的问题。
技术实现要素:
[0016]
为了解决现有技术中替莫唑胺合成路线长,操作繁琐,收率低,纯度低等问题,本发明提供了一种新的替莫唑胺制备方法。该方法反应路线短,操作简便,反应更加温和,产品纯度、收率高,适于工业化生产。
[0017]
本发明具体通过如下技术方案实现:
[0018]
一种替莫唑胺的制备方法,其合成路线如下:
[0019][0020]
一种替莫唑胺的制备方法,具体包括如下步骤:
[0021]
1)将化合物ⅱ加入有机溶剂a中,搅拌溶解,控温加入碱、化合物iii,继续控温反应,反应结束,得中间体化合物iv;
[0022]
2)将中间体化合物iv、亚铜盐、三甲基氰硅烷依次加入有机溶剂b中,控温搅拌反应,反应结束,得中间体化合物
ⅴ
;
[0023]
3)将中间体化合物
ⅴ
加入到酸溶液中,控温水解,反应结束,得化合物i。
[0024]
优选地,步骤1)中所述碱选自二乙基异丙基胺、吡啶、三乙胺中的一种或其组合,其中特别优选二乙基异丙基胺。
[0025]
优选地,步骤1)中所述的有机溶剂a选自甲苯、dmf、dmso中的一种或其组合。
[0026]
优选地,步骤1)中所述的化合物ii、碱、化合物iii的投料摩尔比为1:1.0~4.0:1.0~2.2,其中特别优选1:3.0:2.0。
[0027]
优选地,步骤1)中所述加入碱、化合物iii及反应温度均为-5℃~5℃,其中特别优选0℃。
[0028]
优选地,步骤1)中所述化合物iii的加入方式为将化合物iii溶于有机溶剂a中,然后再滴入反应体系中。
[0029]
在一优选方案中,步骤1)反应结束后需进行后处理操作,具体为:反应结束后,浓缩除去有机相,加入冰水,冰浴降温,搅拌,减压过滤,滤饼依次用冰水、丙酮洗涤,干燥,得中间体化合物iv。
[0030]
优选地,步骤2)中所述亚铜盐选自碘化亚铜、氯化亚铜、溴化亚铜中的一种或其组合,其中特别优选氯化亚铜。
[0031]
优选地,步骤2)中所述中间体化合物iv、无机亚铜盐、三甲基氰硅烷的投料摩尔比为1:0.05~0.1:1.0~2.0,其中特别优选1:0.5:1.2。
[0032]
优选地,步骤2)中所述有机溶剂b选自dmso、n,n-二甲基甲酰胺、n-甲基吡咯烷酮中的一种或其组合,其中特别优选dmso。
[0033]
优选地,步骤2)中所述的反应温度80℃~90℃。
[0034]
在一优选方案中,步骤2)反应结束后需进行后处理操作,具体为:反应结束后加入饱和硫代硫酸钠溶液淬灭,过滤,加入水,然后加入乙酸乙酯萃取,无水硫酸镁干燥,旋蒸,得中间体化合物v。
[0035]
优选地,步骤3)中所述中间体化合物v与酸溶液的投料质量体积比为1:20~50,g/ml。
[0036]
优选地,步骤3)中所述酸溶液选自浓盐酸或浓盐酸与冰乙酸的混酸溶液,其中特别优选浓盐酸与冰乙酸的混酸溶液。
[0037]
进一步优选地,所述的浓盐酸与冰乙酸投料体积比为1:0.2~1.0,其中特别优选1:0.5。
[0038]
优选地,步骤3)中所述水解温度为70℃~75℃。
[0039]
在一优选方案中,步骤3)反应结束后需进行后处理操作,具体为:反应结束后降温搅拌,抽滤,滤饼加入dmso中打浆,抽滤,滤饼用冰乙醇洗涤,干燥,得化合物i。
[0040]
与现有技术相比,本发明提供了一种新的替莫唑胺的制备方法,以化合物ii和甲胺基甲酰氯为原料,廉价易得,整个合成步骤操作简便,反应条件温和,避免了剧毒试剂异氰酸甲酯的使用,相对提高了生产过程中的安全性,经济环保,产品收率及纯度高,适于工业化生产。
具体实施方式
[0041]
下面通过实施例来进一步说明本发明。应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属本发明要求保护的范围。
[0042]
对本发明得到的化合物结构确证:
[0043]
化合物iv的结构表征:
[0044][0045]
化合物iv的高分辨质谱:esi-hrms:m/z=152.0496[m h]
;1h-nmr(400mhz,cdcl3):δ8.15(s,1h),7.03(s,2h),2.75(s,3h);
13
c-nmr(400mhz,cdcl3)δ145.8,139.6,136.7,125.9,37.7.
[0046]
化合物v的结构表征:
[0047][0048]
化合物v的高分辨质谱:esi-hrms:m/z=199.0453[m na]
;1h-nmr(400mhz,cdcl3):δ8.14(s,1h),2.74(s,3h);
13
c-nmr(400mhz,cdcl3):δ145.7,139.7,115.6,136.7,71.4,37.8.
[0049]
中间体化合物iv的制备
[0050]
实施例1
[0051]
将化合物ii(14.11g,0.15mol)加入270ml甲苯中,搅拌溶解,降温至0℃,将二异丙基乙基胺(58.16g,0.45mol)缓慢滴加入反应体系中,15min后将化合物iii(28.05g,0.30mol)溶于50ml甲苯中并缓慢滴加到上述反应体系中,继续控温反应,监测反应完全后,浓缩除去甲苯,加入250ml冰水,搅拌,减压过滤,滤饼用依次用冰水、丙酮洗涤,干燥,得21.42g中间体化合物iv,收率94.5%,hplc纯度99.93%。
[0052]
实施例2
[0053]
将化合物ii(14.11g,0.15mol)加入270ml dmf中,搅拌溶解,降温至5℃,将三乙胺(15.18g,0.15mol)缓慢滴加入反应体系中,15min后将化合物iii(30.86g,0.33mol)溶于
50ml dmf中并缓慢滴加到上述反应体系中,继续控温反应,监测反应完全后,浓缩除去dmf,加入250ml冰水,搅拌,减压过滤,滤饼用依次用冰水、丙酮洗涤,干燥,得20.67g中间体化合物iv,收率91.2%,hplc纯度99.87%。
[0054]
实施例3
[0055]
将化合物ii(14.11g,0.15mol)加入270ml甲苯中,搅拌溶解,降温至-5℃,将吡啶(47.46g,0.60mol)缓慢滴加入反应体系中,15min后将化合物iii(14.03g,0.15mol)溶于50ml甲苯中并缓慢滴加到上述反应体系中,继续控温反应,监测反应完全后,浓缩除去甲苯,加入250ml冰水,搅拌,减压过滤,滤饼用依次用冰水、丙酮洗涤,干燥,得20.56g中间体化合物iv,收率90.7%,hplc纯度99.76%。
[0056]
实施例4
[0057]
将化合物ii(14.11g,0.15mol)加入270ml甲苯中,搅拌溶解,降温至0℃,将三乙胺(30.36g,0.30mol)缓慢滴加入反应体系中,15min后将化合物iii(21.51g,0.23mol)溶于50ml甲苯中并缓慢滴加到上述反应体系中,继续控温反应,监测反应完全后,浓缩除去甲苯,加入250ml冰水,搅拌,减压过滤,滤饼用依次用冰水、丙酮洗涤,干燥,得20.86g中间体化合物iv,收率92.0%,hplc纯度99.90%。
[0058]
实施例5
[0059]
将化合物ii(14.11g,0.15mol)加入270ml甲苯中,搅拌溶解,降温至10℃,将二异丙基乙基胺(58.16g,0.45mol)缓慢滴加入反应体系中,15min后将化合物iii(28.05g,0.30mol)溶于50ml甲苯中并缓慢滴加到上述反应体系中,继续控温反应,监测反应完全后,浓缩除去甲苯,加入250ml冰水,搅拌,减压过滤,滤饼用依次用冰水、丙酮洗涤,干燥,得19.84g中间体化合物iv,收率87.5%,hplc纯度95.69%。
[0060]
实施例6
[0061]
将化合物ii(14.11g,0.15mol)加入270ml dmf中,搅拌溶解,降温至0℃,将二异丙基乙基胺(96.94g,0.75mol)缓慢滴加入反应体系中,15min后将化合物iii(42.08g,0.45mol)溶于50ml dmf中并缓慢滴加到上述反应体系中,继续控温反应,监测反应完全后,浓缩除去dmf,加入250ml冰水,搅拌,减压过滤,滤饼用依次用冰水、丙酮洗涤,干燥,得20.24g中间体化合物iv,收率89.3%,hplc纯度93.74%。
[0062]
实施例7
[0063]
将化合物ii(14.11g,0.15mol)加入270ml甲苯中,搅拌溶解,降温至0℃,将二异丙基乙基胺(38.78g,0.30mol)缓慢滴加入反应体系中,15min后将化合物iii(28.05g,0.30mol)溶于50ml甲苯中并缓慢滴加到上述反应体系中,继续控温反应,监测反应完全后,浓缩除去甲苯,加入250ml冰水,搅拌,减压过滤,滤饼用依次用冰水、丙酮洗涤,干燥,得21.10g中间体化合物iv,收率93.1%,hplc纯度99.91%。
[0064]
中间体化合物v的制备
[0065]
实施例8
[0066]
将中间体化合物iv(15.11g,0.10mol)、氯化亚铜(0.99g,0.01mol)、三甲基氰硅烷(11.90g,0.12mol)依次加入200ml dmso中,85℃搅拌反应,监测反应完全后,加入饱和硫代硫酸钠溶液淬灭,过滤,加入50ml水,然后加入乙酸乙酯(100ml
×
3)萃取,无水硫酸镁干燥,旋蒸,得15.91g中间体化合物v,收率90.3%,hplc纯度99.92%。
[0067]
实施例9
[0068]
将中间体化合物iv(15.11g,0.10mol)、碘化亚铜(0.95g,0.005mol)、三甲基氰硅烷(19.84g,0.20mol)依次加入200ml n,n-二甲基甲酰胺中,80℃搅拌反应,监测反应完全后,加入饱和硫代硫酸钠溶液淬灭,过滤,加入50ml水,然后加入乙酸乙酯(100ml
×
3)萃取,无水硫酸镁干燥,旋蒸,得15.15g中间体化合物v,收率86.0%,hplc纯度99.74%。
[0069]
实施例10
[0070]
将中间体化合物iv(15.11g,0.10mol)、溴化亚铜(2.87g,0.02mol)、三甲基氰硅烷(9.92g,0.10mol)依次加入200ml n-甲基吡咯烷酮中,90℃搅拌反应,监测反应完全后,加入饱和硫代硫酸钠溶液淬灭,过滤,加入50ml水,然后加入乙酸乙酯(100ml
×
3)萃取,无水硫酸镁干燥,旋蒸,得15.34g中间体化合物v,收率87.1%,hplc纯度99.80%。
[0071]
实施例11
[0072]
将中间体化合物iv(15.11g,0.10mol)、碘化亚铜(1.90g,0.01mol)、三甲基氰硅烷(11.90g,0.12mol)依次加入200ml dmso中,90℃搅拌反应,监测反应完全后,加入饱和硫代硫酸钠溶液淬灭,过滤,加入50ml水,然后加入乙酸乙酯(100ml
×
3)萃取,无水硫酸镁干燥,旋蒸,得15.61g中间体化合物v,收率88.6%,hplc纯度99.85%。
[0073]
实施例12
[0074]
将中间体化合物iv(15.11g,0.10mol)、氯化亚铜(0.40g,0.004mol)、三甲基氰硅烷(14.88g,0.15mol)依次加入200ml dmso中,85℃搅拌反应,监测反应完全后,加入饱和硫代硫酸钠溶液淬灭,过滤,加入50ml水,然后加入乙酸乙酯(100ml
×
3)萃取,无水硫酸镁干燥,旋蒸,得14.67g中间体化合物v,收率83.3%,hplc纯度99.67%。
[0075]
实施例13
[0076]
将中间体化合物iv(15.11g,0.10mol)、氯化亚铜(0.99g,0.01mol)、三甲基氰硅烷(11.90g,0.12mol)依次加入200ml thf中,65℃搅拌反应,监测反应完全后,加入饱和硫代硫酸钠溶液淬灭,过滤,加入50ml水,然后加入乙酸乙酯(100ml
×
3)萃取,无水硫酸镁干燥,旋蒸,得14.18g中间体化合物v,收率80.5%,hplc纯度99.42%。
[0077]
替莫唑胺的制备
[0078]
实施例14
[0079]
将中间体化合物v(17.61g,0.10mol)加入至浓盐酸(400ml)与冰乙酸(200ml)的混合溶液中,70℃~75℃控温反应,监测反应完全后,将反应液降温至-5℃,抽滤,滤饼加入dmso(100ml)中打浆,抽滤,滤饼用冰乙醇(25ml
×
2)洗涤,干燥,得19.08g白色偏淡粉固体替莫唑胺,收率98.3%,纯度99.95%。
[0080]
实施例15
[0081]
将中间体化合物v(17.61g,0.10mol)加入至浓盐酸(400ml)中,70℃~75℃控温反应,监测反应完全后,将反应液降温至-5℃,抽滤,滤饼加入dmso(100ml)中打浆,抽滤,滤饼用冰乙醇(25ml
×
2)洗涤,干燥,得18.37g白色偏淡粉固体替莫唑胺,收率94.6%,纯度99.91%。
[0082]
实施例16
[0083]
将中间体化合物v(17.61g,0.10mol)加入至浓盐酸(400ml)与冰乙酸(80ml)的混合溶液中,70℃~75℃控温反应,监测反应完全后,将反应液降温至-5℃,抽滤,滤饼加入
dmso(100ml)中打浆,抽滤,滤饼用冰乙醇(25ml
×
2)洗涤,干燥,得18.66g白色偏淡粉固体替莫唑胺,收率96.1%,纯度99.89%。
[0084]
实施例17
[0085]
将中间体化合物v(17.61g,0.10mol)加入至浓盐酸(400ml)与冰乙酸(400ml)的混合溶液中,70℃~75℃控温反应,监测反应完全后,将反应液降温至-5℃,抽滤,滤饼加入dmso(100ml)中打浆,抽滤,滤饼用冰乙醇(25ml
×
2)洗涤,干燥,得18.83g白色偏淡粉固体替莫唑胺,收率97.0%,纯度99.54%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
