从9-双氢-13-乙酰基巴卡亭iii合成卡巴它赛和多西它赛的方法
技术领域
1.本发明涉及抗癌化合物合成领域,尤其涉及从9-双氢-13-乙酰基巴卡亭iii 合成卡巴它赛和多西它赛的方法。
背景技术:
2.卡巴它赛是“促性腺激素释放激素(gnrh)”受体抑制剂类药物,主要针对晚期前列腺癌患者,是转移性激素难治性前列腺癌的二线治疗中首个及唯一一个提供显著生存获益的治疗药物。美国食品药品管理局批准治疗晚期前列腺癌,临床上被推荐在使用常用晚期前列腺癌药物多烯紫杉醇无效甚至病情加重时,首选的用于治疗晚期、抗激素型前列腺癌的药物。
3.专利cn102532065b,cn102675256a,cn103242267b,us5847170, us2007293687a1,ep0817779,ep0817780,fr2732340,us5889043,us6372780, us6387946,wo9630355,wo9630356等描述了由10-去乙酰基巴卡亭iii(10-dab) 合成卡巴它赛的工艺路线,这些工艺路线都存在工艺复杂,总收率偏低或反应条件要求苛刻难以工业化规模生产;同时10-dab来源于欧洲红豆杉,其资源有限,因此急需扩大卡巴它赛的原料来源及优化其合成工艺。
技术实现要素:
4.本发明克服了现有技术的不足,提供从9-双氢-13-乙酰基巴卡亭iii合成卡巴它赛和多西它赛的方法。
5.为达到上述目的,本发明采用的技术方案为:具有式一所示结构的化合物:
[0006][0007]
其中r1是一个氢原子或合适的羟基保护基。
[0008]
本发明一个较佳实施例中,合适的羟基保护基选自c
1-c
25
醚,c
1-c
25
取代的甲基醚,c
1-c
25
取代的乙基醚,c
1-c
25
酰基,c
1-c
25
卤代酰基,c
1-c
25
苄基醚,c
1-c
25
硅醚,c
1-c
25
酯,c
1-c
25
碳酸酯,c
1-c
25
磺酸酯。
[0009]
本发明一个较佳实施例中,所述合适的羟基保护基选自甲基,甲氧甲基,苄氧甲基,四氢吡喃基,四氢呋喃基,2-(三甲基硅基)乙氧甲基,1-乙氧乙基,1-(2-氯乙氧基)乙基,2,2,2-三氯乙基,叔丁基,烯丙基,炔丙基,苄基,对
ꢀ‑
甲氧基苄基,二苯基甲基,三苯基甲基,三甲基硅基,三乙基硅基,三异丙基硅基,二甲基异丙基硅基,二乙基异丙基硅基,二
甲基甲氧基硅,叔丁基二甲基硅基,叔丁基二苯基硅基,三苄基硅基,三苯基硅基,二苯基甲基硅基,甲基苄酯,甲羰基,乙羰基,甲氧甲羰基,三氯乙氧羰基,苄基羰基,苄氧羰基,烯丙磺酰基,甲基磺酰基,以及对苯基磺酰基。
[0010]
本发明一个较佳实施例中,r1是甲基二苯基硅基。
[0011]
本发明还提供了具有式二所示结构的化合物:
[0012][0013]
其中r1是一个氢原子或合适的羟基保护基;
[0014]
r2是一个氢原子或甲基;
[0015]
r3是甲基或对甲氧基苄基。
[0016]
本发明一个较佳实施例中,合适的羟基保护基选自c
1-c
25
醚,c
1-c
25
取代的甲基醚,c
1-c
25
取代的乙基醚,c
1-c
25
酰基,c
1-c
25
卤代酰基,c
1-c
25
苄基醚,c
1-c
25
硅醚,c
1-c
25
酯,c
1-c
25
碳酸酯,c
1-c
25
磺酸酯。
[0017]
本发明一个较佳实施例中,所述合适的羟基保护基选自甲基,甲氧甲基,苄氧甲基,四氢吡喃基,四氢呋喃基,2-(三甲基硅基)乙氧甲基,1-乙氧乙基, 1-(2-氯乙氧基)乙基,2,2,2-三氯乙基,叔丁基,烯丙基,炔丙基,苄基,对
ꢀ‑
甲氧基苄基,二苯基甲基,三苯基甲基,三甲基硅基,三乙基硅基,三异丙基硅基,二甲基异丙基硅基,二乙基异丙基硅基,二甲基甲氧基硅,叔丁基二甲基硅基,叔丁基二苯基硅基,三苄基硅基,三苯基硅基,二苯基甲基硅基,甲基苄酯,甲羰基,乙羰基,甲氧甲羰基,三氯乙氧羰基,苄基羰基,苄氧羰基,烯丙磺酰基,甲基磺酰基,以及对苯基磺酰基。
[0018]
本发明一个较佳实施例中,r1是甲基二苯基硅基;r2是氢原子;r3是对甲氧基苄基
[0019]
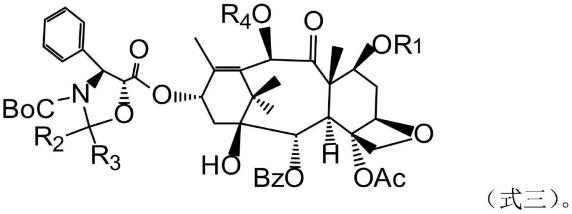
本发明还提供了具有式三所示结构的化合物:
[0020][0021]
本发明还提供了制备式三所示的化合物的过程,包括以下步骤:将氧化剂与有式四所示分子式化合物反应,
[0022][0023]
其中r1是一个氢原子或合适的羟基保护基;
[0024]
r2是一个氢原子或甲基;
[0025]
r3是甲基或对甲氧基苄基;
[0026]
r4是一个氢原子或合适的羟基保护基。
[0027]
本发明一个较佳实施例中,合适的羟基保护基选自c
1-c
25
醚,c
1-c
25
取代的甲基醚,c
1-c
25
取代的乙基醚,c
1-c
25
酰基,c
1-c
25
卤代酰基,c
1-c
25
苄基醚,c
1-c
25
硅醚,c
1-c
25
酯,c
1-c
25
碳酸酯,c
1-c
25
磺酸酯。
[0028]
本发明一个较佳实施例中,所述合适的羟基保护基选自甲基,甲氧甲基,苄氧甲基,四氢吡喃基,四氢呋喃基,2-(三甲基硅基)乙氧甲基,1-乙氧乙基, 1-(2-氯乙氧基)乙基,2,2,2-三氯乙基,叔丁基,烯丙基,炔丙基,苄基,对
ꢀ‑
甲氧基苄基,二苯基甲基,三苯基甲基,三甲基硅基,三乙基硅基,三异丙基硅基,二甲基异丙基硅基,二乙基异丙基硅基,二甲基甲氧基硅,叔丁基二甲基硅基,叔丁基二苯基硅基,三苄基硅基,三苯基硅基,二苯基甲基硅基,甲基苄酯,甲羰基,乙羰基,甲氧甲羰基,三氯乙氧羰基,苄基羰基,苄氧羰基,烯丙磺酰基,甲基磺酰基,以及对苯基磺酰基。
[0029]
本发明一个较佳实施例中,r1是甲基二苯基硅基;r2是一个氢原子或甲基; r3是甲基或对甲氧基苄基;r4是乙酰基。
[0030]
本发明一个较佳实施例中,氧化剂选自戴斯-马丁氧化剂 (dess-martin periodinane),柯林氧化剂(colin’s reagent),斯威氧化剂(swern’s reagent),约翰氧化剂(john’s reagent),氯铬酸吡啶(pcc),重铬酸吡啶盐(pdc),二氧化铬(cro2)和四正丙基过钌酸铵(tpap)。
[0031]
本发明还提供了转换9-双氢-13-乙酰基巴卡亭iii(9-dhab)成为紫杉烷类衍生物的制备过程,包括以下步骤:移除9-双氢-13-乙酰基巴卡亭iii的13位乙酰基及氧化9位羟基。
[0032]
本发明一个较佳实施例中,除去13-位乙酰基是由使用强碱来完成的,包括甲基锂,正丁基锂,红铝,硼氢化钠和水合肼;
[0033]
氧化步骤包括加入下述氧化剂:戴斯-马丁氧化剂(dess-martin periodinane), 柯林氧化剂(colin’s reagent),斯威氧化剂(swern’s reagent),约翰氧化剂 (john’s reagent),氯铬酸吡啶(pcc),重铬酸吡啶盐(pdc),二氧化铬(cro2) 和四正丙基过钌酸铵(tpap)来完成。
[0034]
本发明还提供了卡巴它赛的合成路径,其特征在于,包括以下步骤:
[0035]
(a):脱去9-双氢-13-乙酰基巴卡亭iii的13-位乙酰基以获得9-双氢巴卡亭iii;
[0036]
(b):用合适的保护基保护9-双氢巴卡亭iii的7-位羟基,以便获得一个被保护的
中间体;
[0037]
(c):用合适的氧化剂氧化被保护的中间体的9-位羟基。
[0038]
本发明一个较佳实施例中,保护基包括:苄基,c
1-c
25
取代的苄基,甲酸苄酯,c
1-c
25
取代的甲酸苄酯,苯磺酰基,c
1-c
25
取代的苯磺酰基,甲基,甲氧甲基,c
1-c
25
烷基,c
1-c
25
取代烷基,苯甲酰基,c
1-c
25
取代苯甲酰基,c
1-c
25
三烷基硅基,三甲基硅基,三乙基硅基,三异丙基硅基,二甲基异丙基硅基,二乙基异丙基硅基,二甲基甲氧基硅,叔丁基二甲基硅基,叔丁基二苯基硅基,三苄基硅基,三苯基硅基,二苯基甲基硅基,甲基二苯基硅,苄氧羰基,c
1-c
25
硅醚,c
1-c
25
酯,c
1-c
25
碳酸酯,c
1-c
25
磺酸酯。
[0039]
本发明一个较佳实施例中,氧化剂包括:戴斯-马丁氧化剂 (dess-martin periodinane),柯林氧化剂(colin’s reagent),斯威氧化剂(swern’s reagent),约翰氧化剂(john’s reagent),氯铬酸吡啶(pcc),重铬酸吡啶盐(pdc),二氧化铬(cro2)和四正丙基过钌酸铵(tpap)。
[0040]
本发明一个较佳实施例中,进一步包括脱除中间体7-甲基二苯基硅基巴卡亭 iii-13-【3-叔丁氧羰基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3-噁唑烷】-5-羧酸酯的10
‑ꢀ
位乙酰基的步骤。
[0041]
本发明一个较佳实施例中,脱除10-位乙酰基是由使用强碱来完成,包括正甲醇钠,乙醇钠,丁基锂,甲基锂,红铝,硼氢化钠,和水合肼。
[0042]
本发明一个较佳实施例中,进一步包括加上合适的侧链到7-甲基二苯基硅
ꢀ‑
9-双氢巴卡亭iii的13-位的步骤,以及选择性的脱保护以获得目的化合物。
[0043]
本发明一个较佳实施例中,选择性的脱保护是脱去7-甲基二苯基硅基,10
‑ꢀ
乙酰基以及侧链上的对甲氧基苯甲酰基保护基。
[0044]
本发明一个较佳实施例中,选择性的脱保护是使用碳酸氢钠/过氧化氢混合物以及对甲基苯磺酸来完成的。
[0045]
本发明一个较佳实施例中,所述侧链包括(2r,4s,5r)-3-叔丁氧羰基-2-(4
’ꢀ‑
甲氧基苯基)-4-苯基-1,3-恶唑烷-5-羧酸,(2r,4s,5r)-3-叔丁氧羰基-2-二甲基-4-苯基-1,3-恶唑烷-5-羧酸,(3r,4s)-3-(1-乙氧乙基)-4-苯基-n-叔丁氧羰基-2-氮杂环丁酮,(2r,3s)-n-叔丁氧羰基-o-(1-乙氧乙基)-3-苯基异丝氨酸。
[0046]
本发明一个较佳实施例中,紫杉烷类衍生物是多西它赛和卡巴它赛。
[0047]
本发明解决了背景技术中存在的缺陷,本发明具备以下有益效果:
[0048]
(1)本发明克服了以前的工艺条件反应复杂,试剂毒性大,产品收率低,纯化难度大,生产成本高等不足之处;
[0049]
(2)本发明使用了9-双氢-13-乙酰基巴卡亭iii作为合成开始原料,扩大了卡巴它赛和多西它赛合成的原料来源,简化了工艺;
[0050]
(3)本发明淘汰了传统卡巴它赛和多西它赛合成工艺中使用的高毒性、高污染的化学品,创造性的使用了低毒或无毒性,更为绿色环保的合成试剂;
[0051]
(4)本发明能够获得的产品产率更高,可达到78.0%。
具体实施方式
[0052]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,本发明的描述中,“实施例”、“一个实施例”或“其他实施例”的提及表示结合实施例说明的特定特征、结构或特性包括在至少一些实施例中,但不必是全部实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0053]
实施例一:
[0054]
从9-双氢-13-乙酰基巴卡亭iii合成卡巴它赛的方法,如式六所示,包括以下步骤:
[0055]
s1:在-70℃下,将9-双氢-13-乙酰基巴卡亭iii(9-dhab,100g,158mmol) 溶于2l thf溶液中,随后滴加甲基锂(19.75g ch3li的1.5m thf溶液,790mmol, 5当量)。混合物在-70℃下搅拌3小时,当tlc显示反应已经完成后,反应物用饱和nh4cl溶液淬灭。然后用饱和nacl溶液洗涤有机层,有机相用无水na2so4干燥,过滤后真空浓缩。在真空下除去溶剂后,残余物通过柱层析纯化,得到73g 的9-双氢巴卡亭iii(c1,收率:80%),并回收约10%的9-dhab。1h-nmr(cdcl3) δ8.13(d,2h),7.63(t,1h),7.51(t,2h),6.19(t,1h),5.75(d,1h),5.32(s,1h),4.95(d, 2h),4.81(d,1h),4.48(t,2h),4.34(d,1h),4.19(d,1h),3.77(t,1h),3.13(d,1h),2.55 (ddd,1h),2.33(s,3h,oac),2.20-2.13(m,1h),2.18(s,3h,oac),2.15(s,3h),1.98(m, 1h),1.92(s,3h),1.66(s,3h),1.13(s,3h)ppm。
[0056]
另一实施例中,也可通过以下方式进行纯化:将浓缩至干的残余物加入200 ml ch2cl2,并在室温下搅拌,白色固体开始形成。将混合物在冰水浴中冷却至 2-3℃,并在该温度下放置2小时,白色沉淀通过真空过滤收集,以得到白色固体9-双氢巴卡亭iii(c1),浓缩滤液后残余物用ch3oh结晶,以回收化合物9-dhab。
[0057]
s2:将9-双氢巴卡亭iii(c1,30g,51mmol)加到二氯甲烷(ch2cl2,400ml) 溶液中,搅拌并将此混合物的温度降至0℃。将4,4-二甲基氨基吡啶(dmap,14.3 g,0.1mol,2当量)加入到上述混合物中,随后滴加甲基二苯基氯硅烷(mdpscl, 23.7g,0.1mol,2当量),并在在20分钟内滴完。混合物在0℃下继续搅拌2小时。当tlc显示起始物质消失后,用水淬灭反应混合物。收集有机相,随后用水和盐水洗涤有机层。有机相接着用无水na2so4干燥,过滤并真空浓缩至干。残余物通过dcm/己烷重结晶,过滤收集白色结晶性固体并减压烘干,得到7-甲基二苯基硅基-9-双氢巴卡亭iii(c2,24g,收率80.1%)。1h-nmr(cdcl3)δ8.11 (d,2h),7.77(d,2h),7.62(dd,2h),7.70-7.37(m,9h),6.08(d,1h),5.73(d,1h),5.32 (s,2h),5.16(d,1h),4.78(d,1h),4.77(t,1h),4.58(dd,1h),4.30(t,1h),4.28(d,1h), 4.18(d,1h),3.04(t,1h),2.28-2.16(m,1h),2.25(s,3h,oac),2.19(s,3h,oac),1.97 (s,3h),1.73(s,3h),1.69(s,3h),1.09(s,3h),0.84(s,3h)ppm。
[0058]
可回收约12%的9-甲基二苯基硅基-9-双氢巴卡亭iii。
[0059]
s3:将7-甲基二苯基硅基-9-双氢巴卡亭iii(c2,20.0g,0.15mol)和3-叔丁氧羰基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3-噁唑烷-5-羧酸(多西紫杉醇侧链)(20 g,0.3mol,2当量)加入无水甲苯中的搅拌,随后对此混合溶液中加入4,4-二甲基氨基吡啶(dmap,11g,0.09mol,0.6当量)和二环己基碳二亚胺(dcc, 92.7g,0.45mol,3当量)。将混合物在室温下搅拌2小时。当tlc显示起始物质消失时,用水淬灭反应混合物,然后静置分层。收集有机层并随后用水和盐水洗涤有机层,有机相用无水na2so4干燥,过滤并在真空中浓缩。残余物用硅胶柱层析纯化,得到90%产率的7-甲基二苯基硅基-9-双氢巴卡亭iii-13-【3-叔丁氧羰
基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3-噁唑烷】-5-羧酸酯(c3,21g);1h-nmr(cdcl3) δ8.04(d,2h),7.72(dd,2h),7.62(t,1h),7.59(dd,2h),7.51-7.33(m,13h),6.98(d, 1h),6.03(d,1h),5.69(d,1h),5.32(s,1h),5.17(d,1h),4.65(d,1h),4.58(d,1h), 4.54(dd,1h),4.30(t,1h),4.18(dd,1h),4.15(d,1h),4.09(m,2h),3.86(s,3h),3.85(m, 1h),3.51(d,1h),2.88(d,1h),2.20(m,1h),2.24(s,3h,oac),2.18(s,3h,oac),1.85(s, 3h),1.74(s,3h),1.35(s,3h),1.25(s,3h),1.08(s,9h),0.82(s,3h)ppm。
[0060]
s4:将12g的7-甲基二苯基硅基-9-双氢巴卡亭iii-13-【3-叔丁氧羰基-2-(4
’‑ꢀ
甲氧基苯基)-4-苯基-1,3-噁唑烷】-5-羧酸酯(c3,12g,10mmol)加入100ml的二氯甲烷(ch2cl2)的搅拌溶液中,随后加入戴斯-马丁氧化剂(dess-martin periodinane 12g,25mmol),混合物在室温下搅拌过夜。次日当tlc显示起始物质消失时,用水淬灭反应混合物,收集有机相并用水洗涤有机层,随后用无水 na2so4脱水,过滤并在真空中浓缩。残余物用硅胶柱层析纯化,得到7-甲基二苯基硅基巴卡亭iii-13-【3-叔丁氧羰基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3-噁唑烷】
ꢀ‑
5-羧酸酯(c4,11.2g,收率为92.0%)。1h-nmr(cdcl3)δ8.04(d,2h),7.64(t,1h), 7.55(dd,1h),7.52-7.31(m,15h),6.98(d,1h),6.28(s,1h),5.65(d,1h),4.73(d,1h), 4.58(d,1h),4.44(dd,1h),4.19(dd,1h),4.15(d,1h),4.09(dd,2h),3.87(s,3h),3.66 (d,1h),2.22(m,1h),2.11(s,2h),2.08(s,6h,oac),1.87(s,3h),1.30(s,3h),1.27(s, 3h),1.15(s,3h),1.04(s,9h),0.67(s,3h)ppm。
[0061]
s5:在室温条件下(约25℃),将7-甲基二苯基硅基巴卡亭iii-13-【3-叔丁氧羰基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3-恶唑烷】-5-羧酸酯(c4,7.0g,0.006mol) 加入到20ml thf中并搅拌,随后将nahco3饱和水溶液(7.6g/100ml,15当量) 和h2o2(30%h2o210ml,15当量)添加到该搅拌溶液中。此混合物在25℃下搅拌24小时。当tlc显示起始物质消失时,用水淬灭反应混合物,并用etoac 萃取。有机相用naso3溶液和盐水洗涤,用无水na2so4干燥,过滤并在真空中浓缩。残余物用硅胶柱层析纯化,粗产物在丙酮/正己烷(1:2)中重结晶。10
‑ꢀ
去乙酰基巴卡亭iii-13-【3-叔丁氧羰基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3-噁唑烷-5
‑ꢀ
羧酸酯】(c5,5.3g,收率73%)。
[0062]
s6:将10-去乙酰基巴卡亭iii-13-【3-叔丁氧羰基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3-恶唑烷-5-羧酸酯】(c5,50g,57.5mmol)溶于700ml无水四氢呋喃中(thf),降温至-60℃,随后加入催化剂量的四丁基碘化铵(bu4ni,4.25g,11.5mmol),碳酸二甲酯[(ch3o)2co,26g,287.5mmol,5equiv],搅拌约10分钟后,接着滴入氰化钠(nah,5.5g,230mmol,4equiv)的矿物油溶液,继续低温搅拌一小时随后将温度缓慢升至0℃并维持该温度直到tlc检查反应已完成时停止搅拌。对反应釜中加入300ml纯净水和300ml二氯甲烷的混合溶液继续搅拌5分钟,然后静置分层。收集有机相随后用500ml饱和氯化钠水溶液洗涤,有机相随后减压浓缩至干,残余物用少量丙酮溶解后上反相聚苯乙烯/二乙烯苯聚物材料树脂柱分离纯化,含c6的部分被收集浓缩得白色c6固体32.5g(收率63%)。
[0063]
s7:向c6(5.0g,5.5mmol)在甲醇(50ml)中的溶液中加入p-tsoh(1g, 6mmol),在室温下将混合物搅拌3小时。当tlc显示起始物质消失时,用nahco
3 (0.5g)淬灭反应混合物,在真空中浓缩有机物,并通过柱层析纯化残余物,以 78.0%的产率得到卡巴它赛(cabazitaxel,2.8g)。1h-nmr(400mhz,cdcl3):δ8.10(d,2h),7.65(t,1h),7.52(t,2h),7.30-7.50(m,5h),6.24(t,1h),5.65(d,1h), 5.47(d,1h),4.65(mt,1h),4.33(d,1h),4.20
(d,1h),3.88(dd,1h),3.85(d,1h),3.48(mt, 1h),3.48(s,3h),3.35(s,3h),2.70(m,1h),2.39(s,3h),2.31(m,2h),1.91(s,3h), 1.82(mt,1h),1.75(s,3h),1.70(s,1h),1.39(s,9h),1.25(s,3h),1.23(s,3h)
[0064]
p
[0065][0066]
实施例二:
[0067]
从9-双氢-13-乙酰基巴卡亭iii合成多西它赛的方法,包括以下步骤:
[0068]
步骤a1:在-70℃下,将9-双氢-13-乙酰基巴卡亭iii(9-dhab,100g,158mmol) 溶于2l thf溶液中,随后滴加甲基锂(19.75g ch3li的1.5m thf溶液,790mmol, 5当量)。混合物在-70℃下搅拌3小时,当tlc显示反应已经完成后,反应物用饱和nh4cl溶液淬灭。然后用饱和nacl溶液洗涤有机层,有机相用无水na2so4干燥,过滤后真空浓缩。在真空下除去溶剂后,残余物通过柱层析纯化,得到73g 的9-双氢巴卡亭iii(c1,收率:80%),并回收约10%的9-dhab。
[0069]
另一实施例中,也可通过以下方式进行纯化:将浓缩至干的残余物加入200 ml ch2cl2,并在室温下搅拌,白色固体开始形成。将混合物在冰水浴中冷却至 2-3℃,并在该温度下放置2小时,白色沉淀通过真空过滤收集,以得到白色固体9-双氢巴卡亭iii(c1),浓缩滤液后残余物用ch3oh结晶,以回收化合物9-dhab。
[0070]
步骤a2:将9-双氢巴卡亭iii(c1,30g,51mmol)加到二氯甲烷(ch2cl2, 400ml)溶液中,搅拌并将此混合物的温度降至0℃。将4,4-二甲基氨基吡啶 (dmap,14.3g,0.1mol,2当量)加入到上述混合物中,随后滴加甲基二苯基氯硅烷(mdpscl,23.7g,0.1mol,2当量),并
在在20分钟内滴完。混合物在0℃下继续搅拌2小时。当tlc显示起始物质消失后,用水淬灭反应混合物。收集有机相,随后用水和盐水洗涤有机层。有机相接着用无水na2so4干燥,过滤并真空浓缩至干。残余物通过dcm/己烷重结晶,过滤收集白色结晶性固体并减压烘干,得到7-甲基二苯基硅基-9-双氢巴卡亭iii(c2,24g,收率80.1%)。可回收约12%的9-甲基二苯基硅基-9-双氢巴卡亭iii。
[0071]
步骤a3:将7-甲基二苯基硅基-9-双氢巴卡亭iii(c2,20.0g,0.15mol)和 3-叔丁氧羰基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3-恶唑烷-5-羧酸(多西紫杉醇侧链) (20g,0.3mol,2当量)加入无水甲苯中的搅拌,随后对此混合溶液中加入4,4
‑ꢀ
二甲基氨基吡啶(dmap,11g,0.09mol,0.6当量)和二环己基碳二亚胺(dcc, 92.7g,0.45mol,3当量)。将混合物在室温下搅拌2小时。当tlc显示起始物质消失时,用水淬灭反应混合物,然后静置分层。收集有机层并随后用水和盐水洗涤有机层,有机相用无水na2so4干燥,过滤并在真空中浓缩。残余物用硅胶柱层析纯化,得到90%产率的13-{3-叔丁氧羰基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3
‑ꢀ
恶唑烷-5-羧酸酯基}-7-甲基二苯基硅基-9-双氢巴卡亭iii(c3,21g)。
[0072]
步骤a4:将12g的13-{3-叔丁氧羰基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3-恶唑烷-5-羧酸酯基}-7-甲基二苯基硅基-9-双氢巴卡亭iii(c3,12g,10mmol)加入 100ml的二氯甲烷(ch2cl2)的搅拌溶液中,随后加入戴斯-马丁氧化剂 (dess-martin periodinane 12g,25mmol),混合物在室温下搅拌过夜。次日当 tlc显示起始物质消失时,用水淬灭反应混合物,收集有机相并用水洗涤有机层,随后用无水na2so4脱水,过滤并在真空中浓缩。残余物用硅胶柱层析纯化,得到13-{3-叔丁氧羰基-2-(4
’‑
甲氧基苯基)-4-苯基-1,3-恶唑烷-5-羧酸酯基}-7-甲基二苯基硅基巴卡亭iii(c4,11.2g,收率为92.0%)。
[0073]
步骤a5:向c4(5.0g,5.5mmol)在甲醇(50ml)中的溶液中加入p-tsoh (1g,6mmol),在室温下将混合物搅拌3小时。当tlc显示起始物质消失时,用nahco3(0.5g)淬灭反应混合物,在真空中浓缩有机物,并通过柱层析纯化残余物,得到10-乙酰基多西它赛(c7,2.8g,收率78%)。1h-nmr(cdcl3)δ8.13 (d,2h),7.64(t,1h),7.53(t,2h),7.45-7.33(m,5h),6.92(d,1h),6.31(s,1h),6.26(t, 1h),5.70(d,1h),5.41(t,1h),5.32(s,1h),5.30(t,1h),4.98(d,1h),4.65(brs,1h), 4.44(m,1h),4.34(d,1h),4.19(d,1h),3.83(d,1h),3.39(d,1h),3.34(s,1h),2.57 (ddd,1h),2.53(d,1h),2.40(s,2h),2.34(m,1h),2.27(s,3h,oac),1.95(m,1h),1.87 (s,3h,oac),1.70(s,3h),1.65(s,3h),1.36(s,9h),1.27(s,3h),1.18(s,3h)ppm。
[0074]
步骤a6:进行如式七所示的反应,从而得到多西它赛。在室温条件下(约 25℃),将10-乙酰基多西它赛(c7,7.0g,0.006mol)加入到20ml thf中并搅拌,随后将nahco3饱和水溶液(7.6g/100ml,15当量)和h2o2(30%h2o210ml,15 当量)添加到该搅拌溶液中。此混合物在25℃下搅拌24小时。当tlc显示起始物质消失时,用水淬灭反应混合物,并用etoac萃取。有机相用naso3溶液和盐水洗涤,用无水na2so4干燥,过滤并在真空中浓缩。残渣通过快速色谱纯化,得到粗多西它赛,粗产物用丙酮/正己烷=1/2重结晶,得到多西它赛白色结晶 (5.3g,收率73%)。1h-nmr(cdcl3)δ8.13(d,2h),7.66(t,1h),7.42(t,2h),7.36(dd, 4h),7.34(t,1h),6.24(t,1h),5.71(d,1h),5.45(d,1h),5.29(d,1h),5.23(s,1h),4.98 (d,1h),4.64(brs,1h),4.35(d,1h),4.27(m,1h),4.23(s,1h),4.21(d,1h),3.95(d, 1h),3.39(d,1h),2.62(ddd,1h),2.40(s,3h),2.30(d,1h),1.90(dd,1h),1.89(s,3h, oac),1.84(s,
3h),1.79(s,1h),1.56(d,1h),1.37(s,9h),1.27(s,3h),1.16(s,3h)ppm。
[0075][0076]
以上依据本发明的理想实施例为启示,通过上述的说明内容,相关人员完全可以在不偏离本项发明技术思想的范围内,进行多样的变更以及修改。本项发明的技术性范围并不局限于说明书上的内容,必须要根据权利要求范围来确定技术性范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。