一种通过铑金属催化偶联的方式合成
α
,
β
–
不饱和酮方法
技术领域
1.本发明属有机化学领域,具体涉及一种合成α,β
–
不饱和酮的方法。
背景技术:
2.α,β
–
不饱和酮类化合物在自然界中广泛存在,它在生物医药,有机化学合成等诸多领域都有广泛的应用前景。合成α,β
–
不饱和酮的方法也是多种多样的,如硅基烯醇醚的脱氢硅化反应, homer
‑
wadsworth
‑
emmons反应/witting反应使用磷试剂作为底物,金属催化氧化反应和过渡金属交叉偶联反应等。然而,这些合成α,β
–
不饱和酮的方法都存在一定的缺点,如碱性或酸性反应条件要求强,产物收率低,反应条件苛刻,反应时间长,使用有毒试剂,区域选择性低等缺点。本方法使用过渡金属铑作为催化剂,使用烯基硅和羧酸作为原料,有效的合成了具有单一构型的α,β
–
不饱和酮类化合物,且该方法使用的试剂稳定性高,反应过程中不需要强酸强碱的参与,反应产率及反应适应范围广泛等优点。
技术实现要素:
3.本发明的目的在于提供一种有效的合成α,β
–
不饱和酮的方法,该方法经过金属铑催化偶联的方式,解决传统合成方法中,反应过程需加入强酸强碱类试剂,产物的产率低以及化学选择性低等缺点。
4.为了解决本发明的技术问题,所提出的技术方案为:一种通过铑金属催化偶联的方式合成α,β
–
不饱和酮方法,包括以下步骤:
5.步骤一:将烯基硅衍生物(式一),羧酸类衍生物(式2),六水合氯化镁,金属催化剂四羰基二氯化二铑和六水合氯化镁,二碳酸二叔丁酯加进放有磁子的干燥瓶子,并且给两口瓶装冷凝管;
6.步骤二,在氮气氛围下缓慢加入有机溶剂,通冷凝水,在80
‑
110℃下搅拌反应,待烯基硅衍生物完全反应,停止反应,冷却至室温;
7.步骤三,将在步骤二中得到的混合物,进行过滤,洗涤,再将得到的滤液用旋转蒸发仪在低压下旋干,最后用硅胶柱对得到的粗产品进行分离提纯,得到相应的α,β
–
不饱和酮衍生物(式三);
8.具体反应方程式如下:
[0009][0010]
其中r1为:烷基或苯基;r2为:烷基或苯基;r3为:烷基或苯基;r4为烷基或苯基,r5为烷基或苯基。
[0011]
优选的,所述羧酸类衍生物选自脂肪酸、芳香族羧酸或杂环类羧酸。
[0012]
优选的,所述脂肪酸选自甲酸,乙酸,丙酸,丁酸,异丁酸,叔丁酸,环己酸或环戊
酸。
[0013]
优选的,所述芳香族羧酸选自苯甲酸,苯乙酸,2
‑
萘酸,对甲氧基苯甲酸或对甲氧基苯乙酸。
[0014]
优选的,所述烯基硅类衍生物选自脂肪族、芳香族或杂环类的烯基硅。
[0015]
优选的,所述反应的温度为100℃;所述反应的时间为6h~24h。
[0016]
优选的,所述反应摩尔比为烯基硅衍生物:羧酸类衍生物:碳酸二叔丁酯:四羰基二氯化二铑:六水合氯化镁=1:4:3:0.05:0.1。
[0017]
优选的,所述溶剂选自苯或甲苯。
[0018]
本发明的具体实验方案如下:
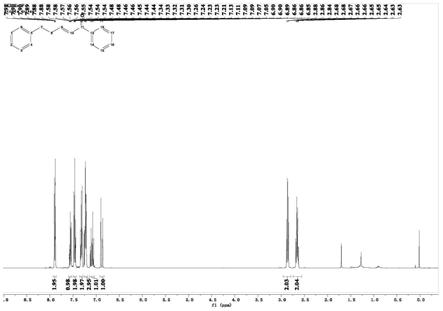
[0019]
铑催化的合成α,β
–
不饱和酮类化合物的制备方案如下:
[0020]
步骤一:将烯基硅衍生物1式一(1equiv,0.1mmol),羧酸2式二(4equiv,0.4mmol),六水合氯化镁(20mmol%),四羰基二氯化二铑([rhcl(co)2]2)(5mmol%)和二碳酸二叔丁酯((t
‑
buoco2)2o) (3equiv,0.3mmol)加进放有磁子的干燥25ml两口瓶中,并且给两口瓶装冷凝管;
[0021][0022]
步骤二:在氮气氛围下缓慢加入2ml有机溶剂苯,通冷凝水,在 100℃下搅拌反应并通过tlc板监测反应,待烯基硅衍生物完全反应,停止反应,冷却至室温;
[0023]
步骤三,将在步骤二中得到的混合物,进行过滤,洗涤,再将得到的滤液用旋转蒸发仪在低压下旋干,最后用硅胶柱对得到的粗产品进行分离提纯,得到相应的α,β
–
不饱和酮类化合物如式三所示。
[0024][0025]
式一,式二,式三中r1为:烷基,苯基,杂环,取代苯基,苯并杂环等。
[0026]
式一,式二,式三中r2为:烷基,苯基,取代苯基,杂环等
[0027]
式一中r3,r4,r5为甲基或苯基
[0028]
步骤二中tlc板监测反应,反应时间为7
‑
24h.
[0029]
步骤三中得到的α,β
–
不饱和酮类化合物为单一的(e)式构型的产物
[0030]
步骤一中提到的四羰基二氯化二铑的摩尔量为5mmol%
[0031]
步骤一中提到的四羰基二氯化二铑的摩尔量为10mmol%
[0032]
步骤二中所用到的最优溶剂为苯,但不仅限于此一溶剂。
[0033]
与现有技术相比,本发明的有益效果:
[0034]
1.本发明的反应体系简单,反应中只需加入催化剂,所选用的底物稳定易得,制备
成本低;
[0035]
2.本发明的反应体系干净,转化率较高,仅需将体系中的催化剂过滤掉即可,产物易分离;
[0036]
3.本发明所用方法适用性广泛,大多数常见的羧酸的均适用本方法,底物的适用性广泛,按照此方法可制备一系列α,β
–
不饱和酮类化合物。
[0037]
4.本发明所用到的金属催化剂四羰基二氯化二铑和六水合氯化镁是在筛选条件后的得到的具有唯一性的可以催化该反应的催化剂。
[0038]
5.本发明的添加剂(t
‑
buoco2)2o在本反应中的主要作用是促使羧酸生成酸酐,且经过筛选证明,在本反应中具有唯一性。
附图说明
[0039]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
[0040]
图1是本发明实施例1提供的(e)
‑
1,5二苯基戊
‑2‑
烯
‑1‑
酮的核磁共振1h谱图;
[0041]
图2是本发明实施例1提供的(e)
‑
1,5二苯基戊
‑2‑
烯
‑1‑
酮的核磁共振
13
c谱图;
[0042]
图3是本发明实施例1提供的(e)
‑
查尔酮的核磁共振1h谱图;
[0043]
图4是本发明实施例1提供的(e)
‑
查尔酮的核磁共振
13
c谱图
[0044]
图5是本发明实施例(e)
‑
6苯基己
‑3‑
烯
‑2‑
酮的核磁共振1h 谱图;
[0045]
图6是本发明实施例(e)
‑
6苯基己
‑3‑
烯
‑2‑
酮的核磁共振
13
c 谱图。
具体实施方式
[0046]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0047]
本发明的方法根据反应底物结构的不同,通过同一个机理,可合成各种类型的α,β
–
不饱和酮类化合物。
[0048]
(1)以(e)
‑
二甲基(苯基)(4
‑
苯基丁
‑1‑
烯)硅烷作为模板底物,使用不同种类的羧酸化合物来讨论方法的实用性。具体反应方程式如下:
[0049][0050]
其中,r为苯基,对甲氧基苯基,萘基,对甲氧基苄基,环丙基,环戊基,甲基,丁基,叔丁基,2
‑
四氢呋喃基,2
‑
呋喃基,2
‑
噻吩基等。其结构式如下:
[0051][0052]
(2)以苯甲酸作为模板底物,使用不同种类的烯基硅类化合物来讨论方法的实用性。具体反应方程式如下:
[0053][0054]
其中r2为甲基或苯基,但不限于甲基或苯基。r基为1
‑
苯基
‑
丙烷,苯基,环戊基,环己基,叔丁基,环戊烯,环己烯,,呋喃,苯丙呋喃等。具体结构如下:
[0055][0056]
实施例1
[0057]
本实施例进行(e)
‑
1,5二苯基戊
‑2‑
烯
‑1‑
酮的制备。具体操作如下:
[0058][0059]
在氮气环境下,(e)
‑
二甲基(苯基)(4
‑
苯基丁
‑1‑
烯基)硅烷(0.1 mmol),苯甲酸(0.4mmol),二碳酸二叔丁酯(0.3mmol),mgcl2.6h2o (10mmol%),[rhcl(co)2]2(5mmol%)和苯(2ml)加进装有冷凝管的25ml的两口瓶中。在100℃下反应,使用tlc板监测反应,待到(e)
‑
二甲基(苯基)(4
‑
苯基丁
‑1‑
烯)硅烷完全反应,停止加热并将反应冷却至室温。将得到的混合物经过砂芯漏斗过滤,用二氯甲烷洗涤(10ml, 3
‑
5次),然后将溶剂用旋转蒸发仪在低压下蒸发,得到粗产品。粗产品经硅胶柱层析(2%ea/pe)纯化,得到所需产品(e)
‑
1,5二苯基戊
ꢀ‑2‑
烯
‑1‑
酮,产物为无色油状液体,产率为73%(17.3mg)。
[0060]
对(e)
‑
1,5二苯基戊
‑2‑
烯
‑1‑
酮核磁共振氢谱的测定,结果为:1h nmr(400mhz,chloroform
‑
d)δ7.92
–
7.86(m,2h),7.59
–
7.53(m, 1h),7.49
–
7.43(m,2h),7.36
–
7.28(m,2h),7.25
–
7.19(m,3h),7.09 (dt,j=15.4,6.8hz,1h),6.88(dt,j=15.4,1.4hz,1h),2.86(t,j=7.7 hz,2h),2.74
–
2.56(m,2h).
[0061]
对(e)
‑
1,5二苯基戊
‑2‑
烯
‑1‑
酮核磁共振碳谱的测定,结果为:
13
c nmr(101mhz,chloroform
‑
d)δ190.98,148.57,140.92,137.96, 132.78,128.66,128.62,128.52,126.65,126.31,34.64,34.59.
[0062]
实施例2
[0063]
本实施例进行(e)
‑
查尔酮的制备。具体操作如下:
[0064][0065]
在氮气环境下,(e)
‑
二甲基(苯基)(苯乙烯基)硅烷(0.1mmol), 苯甲酸(0.4mmol),二碳酸二叔丁酯(0.3mmol),mgcl2.6h2o(10 mmol%),[rhcl(co)2]2(5mmol%)和苯(2ml)加进装有冷凝管的25ml的两口瓶中。在100℃下反应,使用tlc板监测反应,待到(e)
‑
二甲基(苯基)(苯乙烯基)硅烷完全反应,停止加热并将反应冷却至室温。将得到的混合物经过砂芯漏斗过滤,用二氯甲烷洗涤(10ml,3
‑
5 次),然后将溶剂用旋转蒸发仪在低压下蒸发,得到粗产品。粗产品经硅胶柱层析(2%ea/pe)纯化,得到所需产品(e)
‑
查尔酮,产物为无色油状液体,产率为60%(12.5mg)。
[0066]
对(e)
‑
查尔酮核磁共振氢谱的测定,结果为:1h nmr(400mhz, chloroform
‑
d)δ8.06
–
7.99(m,2h),7.82(d,j=15.7hz,1h),7.70
–ꢀ
7.48(m,6h),7.46
–
7.38(m,3h).
[0067]
对(e)
‑
查尔酮核磁共振碳谱的测定,结果为:
13
c nmr(101mhz, chloroform
‑
d)δ190.74,145.01,138.33,135.01,132.94,130.71,129.11, 128.78,128.65,128.60,122.21.
[0068]
实施例3
[0069]
本实施例进行(e)
‑
6苯基己
‑3‑
烯
‑2‑
酮的制备。具体操作如下:
[0070][0071]
在氮气环境下,(e)
‑
二甲基(苯基)(4
‑
苯基丁
‑1‑
烯)硅烷(0.1 mmol),乙酸(0.4mmol),二碳酸二叔丁酯(0.3mmol),mgcl2.6h2o (10mmol%), [rhcl(co)2]2(5mmol%)和苯(2ml)加进装有冷凝管的25ml的两口瓶中。在100℃下反应,使用tlc板监测反应,待到(e)
‑
二甲基(苯基)(4
‑
苯基丁
‑1‑
烯)硅烷完全反应,停止加热并将反应冷却至室温。将得到的混合物经过砂芯漏斗过滤,用二氯甲烷洗涤(10ml, 3
‑
5次),然后将溶剂用旋转蒸发仪在低压下蒸发,得到粗产品。粗产品经硅胶柱层析(2%ea/pe)纯化,得到所需产品(e)
‑
6苯基己
‑3‑ꢀ
烯
‑2‑
酮,产物为无色油状液体,产率为79%(13.8mg)。
[0072]
对(e)
‑
6苯基己
‑3‑
烯
‑2‑
酮核磁共振氢谱的测定,结果为:1h nmr(400mhz,chloroform
‑
d)δ7.34
–
7.27(m,2h),7.25
–
7.16(m, 3h),6.82(dt,j=15.9,6.8hz,1h),6.10(d,j=16.0hz,1h),2.87
–
2.71 (m,2h),2.65
–
2.46(m,2h),2.23(s,3h).
[0073]
对(e)
‑
6苯基己
‑3‑
烯
‑2‑
酮核磁共振碳谱的测定,结果为:13c nmr(101mhz,chloroform
‑
d)δ198.84,147.33,131.78,128.62,128.42, 126.34,34.48,34.25,27.01.
[0074]
对比列1
[0075]
本对比列与实施列1(e)
‑
1,5二苯基戊
‑2‑
烯
‑1‑
酮的制备进行对比,具体操作步骤如下:
[0076][0077]
在氮气环境下,(e)
‑
二甲基(苯基)(4
‑
苯基丁
‑1‑
烯基)硅烷(0.1 mmol),苯甲酸(0.4mmol),二碳酸二叔丁酯(0.3mmol),mgcl2.6h2o (10mmol%),[rhoh(cod)2]2(5mmol%)和苯(2ml)加进装有冷凝管的25ml的两口瓶中。在100℃下反应,反应12h后,发现没有任何产物生成,且(e)
‑
二甲基(苯基)(4
‑
苯基丁
‑1‑
烯基)硅烷没有发生任何转化。
[0078]
对比列2
[0079]
本对比列与实施列1(e)
‑
1,5二苯基戊
‑2‑
烯
‑1‑
酮的制备进行对比,具体操作步骤如下:
[0080][0081]
在氮气环境下,(e)
‑
二甲基(苯基)(4
‑
苯基丁
‑1‑
烯基)硅烷(0.1 mmol),苯甲酸(0.4mmol),mgcl2.6h2o(10mmol%),[rhcl(co)2]
2 (5mmol%)和苯(2ml)加进装有冷凝管的25ml的两口瓶中。在 100℃下反应,反应12h后,发现没有任何产物生成,且(e)
‑
二甲基 (苯基)(4
‑
苯基丁
‑1‑
烯基)硅烷没有发生任何转化。
[0082]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。