1.本发明属于生物医药领域,具体涉及一种鉴别不同人群的方法。
背景技术:
2.大多数肠道微生物对人体生理和健康都有着重要影响,它们对人类生命至关重要。肠道菌群与肠道细胞之间的相互作用可以调节屏障功能,不断刺激免疫系统防御病原体。肠道菌群与宿主之间的平衡如果发生了改变,可能会导致机体产生各种疾病。高海拔地区缺氧环境的人群成功地在极端环境条件下逐代繁衍,因此其对高海拔地区缺氧环境的快速适应机制也一直是科学家们关注的热点问题。长期生活在极端环境中的人群独特的饮食习惯和生活方式使得他们的肠道菌群具有独特结构组成,研究菌群的特殊结构与特定人群的疾病的关系,有利于探讨高原疾病发生机理。
技术实现要素:
3.本发明的第一目的在于提供微生物在鉴别或区分不同人群中的应用;
4.本发明的第二目的在于提供一种鉴别或区分不同人群的方法。
5.为实现上述目的,本发明采用了如下技术方案:
6.本发明第一方面提供了一种可用于鉴别或区分不同人群的试剂,所述的试剂能够检测微生物的丰度水平,所述的微生物包括s_actinobacillussuccinogenes、s_actinomyces sp.hmsc035g02、s_alistipes indistinctus、 s_citrobacter sp.mgh110、s_clostridium sp.cag_58、s_cohnella sp.ov330、 s_fibrobacter sp.uwov1、s_klebsiella sp.obrc7、s_klebsiella variicola、 s_kluyvera ascorbata、s_kytococcus sedentarius、s_lactobacilluskefiranofaciens、s_lactococcus garvieae、s_lysobacter enzymogenes、 s_olsenella umbonata、s_paenibacillus massiliensis、s_pantoea ananatis、s_peptostreptococcus sp.d1、s_porphyromonas sp.hmsc065f10、s_prevotellaaurantiaca、s_slackia piriformis、s_candidatus saccharibacteria oral taxontm7x和/或s_lachnospiraceae bacterium khcpx20。
7.术语“和/或”是指并且包括一个或多个相关联的所列项目的任何和所有可能的组合,以及在备选方案(或)中解释时缺少组合。
8.术语“丰度”是指生物样品中目标微生物的数量的量度。“丰度”也被称为“负载”。一般通过分子方法,典型地通过例如荧光原位杂交(fish)、定量聚合酶链反应(qpcr)或pcr/焦磷酸测序测定所述的目标微生物的16s rrna基因拷贝数,进行细菌定量。生物样品内目标核酸序列丰度的定量可能是绝对的或相对的。“相对定量”通常是基于一个或多个内部参考基因,即来自参考菌株的16s rrna基因,比如使用通用引物并且将目标核酸序列的丰度表达为总细菌16s rrna基因拷贝的百分比或通过大肠杆菌16s rrna基因拷贝归一化而测定的细菌。“绝对定量”通过与dna标准进行比较或通过dna浓度归一化来给出目标分子的确切数目。
9.本文所使用的术语“生物样品”指的是从患者处获得的流体样品、细胞样品、组织样品或器官样品。在一些实施方式中,从受试者处获得细胞或细胞群、或一定量的组织或体液。“生物样品”经常可包括来自动物的细胞,但该术语也可以指非细胞的生物材料,如可用于检测微生物的存在或类别的血液、唾液或尿液的非细胞部分。生物样品包括但不仅限于:活组织切片、刮取物(如口腔刮取物)、全血、血浆、血清、尿液、唾液、细胞培养物、活组织切片、粘膜样品、粪便、肠灌洗物、关节液、脑脊液、胆汁样品、呼吸道分泌物(如痰)、支气管肺泡灌洗液样品等。生物样品或组织样品可以指由个体分离的组织或流体,包括但不仅限于,例如,血、血浆、血清、尿、粪便、痰、脊髓液、胸膜液、淋巴液;皮肤、呼吸道、肠道和泌尿生殖道的外层;眼泪、唾液;和器官。样品可包括冷冻组织。术语“样品”还涵盖任何由对此类样品进行进一步加工而衍生的材料。衍生样品可包括例如由样品提取的核酸或蛋白;或经由将所述样品进行如核酸扩增或 mrna逆转录,或对特定核酸、蛋白、其它细胞质组分或核组分进行分离和/或纯化等技术而获得的核酸或蛋白。
10.进一步,所述的试剂包括引物、探针、反义寡核苷酸、适配体或抗体。
11.术语“引物”指的是能够形成与模板链互补的碱基对(bas e pair),并且起到用于复制模板链的起始点作用的7个~50个核酸序列。引物通常合成而得,但也可以使用自然生成的核酸。引物的序列并不一定需要与模板的序列完全相同,只要充分互补而能够与模板杂交即可。可以混入不改变引物的基本性质的追加特征。作为可以混入的追加特征的例子,有甲基化、带帽、一个以上的核酸被同系物取代和核酸间的修饰,但不限于此。
12.术语“杂交”指的是两个互补的核酸链在适当严格的条件下彼此退火结合。通常利用探针长度的核酸分子来进行杂交。核酸杂交技术在现有技术中是公知的。本领域的技术人员了解如何估计和调整杂交条件的严格度,使得具有至少所需程度的互补性的序列将稳定地杂交,而具有较低互补性的序列将不能稳定地杂交。
13.术语“探针”指的是能与另一分子的特定序列或亚序列或其它部分结合的分子。除非另有指出,术语“探针”通常指能通过互补碱基配对与另一多核苷酸(往往称为“靶多核苷酸”)结合的多核苷酸探针。根据杂交条件的严谨性,探针能和与该探针缺乏完全序列互补性的靶多核苷酸结合。探针可作直接或间接的标记,其范围包括引物。杂交方式包括,但不限于:溶液相、固相、混合相或原位杂交测定法。
14.术语“寡核苷酸”指的是由脱氧核糖核苷酸、核糖核苷酸或其任意组合构成的短聚合物。寡核苷酸的长度通常在大10个核苷酸和大约100个核苷酸之间。寡核苷酸优选地长度为15个核苷酸到70个核苷酸,最通常的是20个核苷酸到26个核苷酸。寡核苷酸可以用作引物或探针。
15.术语“适配体”是通过链内碱基间的氢键作用折叠形成稳定的发卡、茎环、假结、口袋、凸环和g-四链体等二级或三级结构,并与靶标产生空间结构匹配的高亲和力和特异性结合的核糖核酸和单链脱氧核糖核酸。
16.在本发明中,术语“抗体”以最广义使用,而且具体涵盖例如单克隆抗体,多克隆抗体,具有多表位特异性的抗体,单链抗体,多特异性抗体和抗体片段。此类抗体可以是嵌合的,人源化的,人的和合成的。
17.本发明第二方面提供了本发明第一方面所述的试剂在制备用于鉴别或区分不同人群的产品中的应用。
18.进一步,所述的产品包括试剂盒、芯片或高通量测序平台。
19.术语“芯片”可指具有附着有吸附剂的、一般为平面的表面的固体基底。生物芯片的表面可包含多个可寻址的位置,其中每个位置可结合有吸附剂。生物芯片可适合于接合探针接口,并因此用作探针。蛋白质生物芯片适用于捕获多肽,并可包含在可寻址位置处附着有层析或生物特异性吸附剂的表面。微阵列芯片一般用于dna和rna基因表达检测。
20.进一步,所述的不同人群包括大众人群、特定人群。
21.本发明第三方面提供了微生物在构建不同人群的分类模型中的应用,所述的微生物包括s_actinobacillus succinogenes、s_actinomyces sp. hmsc035g02、s_alistipes indistinctus、s_citrobacter sp.mgh110、 s_clostridium sp.cag_58、s_cohnella sp.ov330、s_fibrobacter sp.uwov1、 s_klebsiella sp.obrc7、s_klebsiella variicola、s_kluyvera ascorbata、 s_kytococcus sedentarius、s_lactobacillus kefiranofaciens、s_lactococcusgarvieae、s_lysobacter enzymogenes、s_olsenella umbonata、s_paenibacillusmassiliensis、s_pantoea ananatis、s_peptostreptococcus sp.d1、 s_porphyromonas sp.hmsc065f10、s_prevotella aurantiaca、s_slackiapiriformis、s_candidatus saccharibacteria oral taxon tm7x和/或 s_lachnospiraceae bacterium khcpx20。
22.进一步,所述的不同人群的分类模型使用选自以下一种或更多种算法来确定:xgboost、随机森林、glmnet、cforest、机器学习的分类与回归树、 treebag、k-毗邻、神经网络、支持向量机径向、支持向量机线性、朴素贝叶斯或多层感知。
23.在本发明中,“模型”是任何数学方程式,算法,分析或程序化过程或统计技术,其采用一个或多个连续或分类输入并计算输出值,有时称为“索引”,“索引值”,“预测器”,“预测值”,“概率”或“概率得分”。“公式”的非限制性示例包括和、比率以及回归算子,例如系数或指数,生物标志物值转换和标准化,规则和指南,统计分类模型以及对历史群体进行训练的神经网络。在组(panel)和组合构造中,特别有趣的是结构和句法统计分类算法,以及利用模式识别特征的风险指数构建方法,包括已建立的技术,例如互相关,主成分分析(pca),因子旋转,对数回归(logreg),线性判别分析(lda),特征基因线性判别分析(eigengene lineardiscriminantanalysis,elda),支持向量机(support vector machines,svm),随机森林 (random forest,rf),递归分区树(rpart)、xgboost(xgb)以及其他相关的决策树分类技术,shrunkencentroids(sc),stepaic,最近的kth邻居 (kth-nearest neighbor),boosting,决策树(decision trees),神经网络,贝叶斯网络,支持向量机和隐马尔可夫模型(hidden markovmodels)等。还进一步实现了许多此类算法技术,以执行特征(基因座)选择和规则化 (regularization)规则化,例如在岭回归(ridge regression),lasso和elastic net 等中。其他技术可用于生存和事件前时间危险分析(time to event hazardanalysis)中,包括本领域技术人员众所周知的cox,weibull,kaplan-meier 和greenwood模型。这些技术中的许多技术都可以与生物标志物选择技术结合使用,例如正向选择,后向选择或逐步选择,给定大小的所有潜在生物标志物集或组的完整枚举,遗传算法或它们本身可以包括生物标志物选择方法。这些可以与信息标准结合使用,例如akaike的信息标准 (akaike'sinformation criterion,aic)或贝叶斯信息标准(bayes informationcriterion,bic),以便量化其他生物标志物和模型改进之间的权衡,并有助于最小化过度
拟合。生成的预测模型可以在其他研究中进行验证,或在它们最初进行培训的研究中交叉验证,使用诸如bootstrap, leave-one-out(loo)和10倍交叉验证(10-fold cross-validation)(10倍cv) 等技术进行。在各个步骤,可以根据本领域已知的技术通过值排列来估计错误发现率。
24.本发明第四方面提供了一种鉴别或区分不同人群的方法,所述的方法包括检测微生物的丰度水平,所述的微生物包括s_actinobacillus succinogenes、 s_actinomyces sp.hmsc035g02、s_alistipes indistinctus、s_citrobacter sp. mgh110、s_clostridium sp.cag_58、s_cohnella sp.ov330、s_fibrobacter sp. uwov1、s_klebsiella sp.obrc7、s_klebsiella variicola、s_kluyvera ascorbata、s_kytococcus sedentarius、s_lactobacillus kefiranofaciens、s_lactococcusgarvieae、s_lysobacter enzymogenes、s_olsenella umbonata、s_paenibacillusmassiliensis、s_pantoea ananatis、s_peptostreptococcus sp.d1、 s_porphyromonas sp.hmsc065f10、s_prevotella aurantiaca、s_slackiapiriformis、s_candidatus saccharibacteria oral taxon tm7x和/或 s_lachnospiraceae bacterium khcpx20。
25.进一步,所述的检测受试者样本中微生物的丰度水平通过以下任意一种或多种方法来实现:16s rrna测序、全基因组测序、定量聚合酶链反应、 pcr-焦磷酸测序、荧光原位杂交、微阵列、pcr-elisa。
26.术语“测序”是指测定核酸分子(例如,dna或rna核酸分子)中的核苷酸碱基——a、t、c、g和u——的顺序的测序方法。
27.术语“宏基因组”("metagenome")涉及包括在诸如土壤、动物肠等分离的区域中的所有病毒、细菌、真菌等的全部基因组,并且主要用作基因组的概念,其解释了使用测序仪一次鉴定许多微生物以分析非培养的微生物。特别地,宏基因组不是指一种物种的基因组,而是指基因组的混合物,包括环境单位的所有物种的基因组。这个术语源于这样一种观点:当在生物学发展到组学(omics)的过程中定义一个物种时,各种物种以及现有的一个物种在功能上相互作用以形成完整的物种。在技术上,它是使用快速测序以识别一个环境中的所有物种并验证相互作用和代谢来分析所有dna和 rna的技术的主题,无论物种如何都如此。
28.术语“核酸”泛指:染色体的段;dna、cdna和/或rna的段或部分。核酸可以自最初与任何源分离的核酸样本(例如,与样本dna或rna分离、从样本dna或rna纯化、扩增、克隆或逆转录)获取或获得。
29.术语“16s”、“16s核糖体亚基”和“16s核糖体rna(rrna)”可在本文中互换使用,并且可指原核生物(例如细菌、古细菌)核糖体小亚基(例如30s) 的组分。16s rrna在微生物物种之间在进化上是高度保守的。因此,16s 核糖体亚基的测序可用于鉴定和/或比较样品中存在的微生物(例如微生物组)。
30.本发明第五方面提供了一种鉴别或区分不同人群的系统,包括以下单元:
31.1)检测单元:包括微生物检测模块;
32.2)分析单元:将检测单元检测得到的微生物的丰度水平作为输入变量,输入不同人群的分类模型进行分析;
33.3)评估单元:输出样本对应的个体为大众人群/特定人群的概率值;
34.所述的微生物包括s_actinobacillus succinogenes、s_actinomyces sp. hmsc035g02、s_alistipes indistinctus、s_citrobacter sp.mgh110、 s_clostridium sp.cag_58、s_cohnella sp.ov330、s_fibrobacter sp.uwov1、 s_klebsiella sp.obrc7、s_klebsiella variicola、s_kluyvera ascorbata、 s_kytococcus sedentarius、s_lactobacillus kefiranofaciens、s_lactococcusgarvieae、s_lysobacter enzymogenes、s_olsenella umbonata、s_paenibacillusmassiliensis、s_pantoea ananatis、s_peptostreptococcus sp.d1、 s_porphyromonas sp.hmsc065f10、s_prevotella aurantiaca、s_slackiapiriformis、s_candidatus saccharibacteria oral taxon tm7x和/或 s_lachnospiraceae bacterium khcpx20。
附图说明
35.图1为每个特征的贡献值结果图;
36.图2为特征数目与auc值对应关系图;
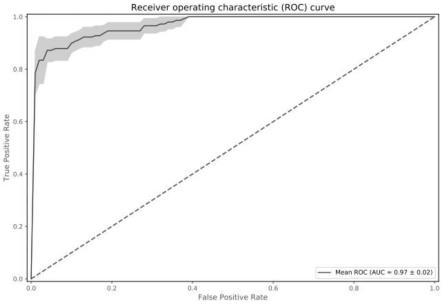
37.图3为最优模型的roc曲线。
具体实施方式
38.下面结合附图和实施例对本发明作进一步详细的说明。以下实施例仅用于说明本发明而不用于限制本发明的范围。实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。
39.实施例1大众人群和特定人群的分类模型
40.一、人群信息
41.大众人群:大众人群1(182人);大众人群2(92人);大众人群3(85 人);大众人群4(63人);大众人群5(143人);大众人群6(43人);大众人群7(30人);
42.特定人群(138人)。
43.二、实验方法
44.1、粪便样本采集与dna提取
45.收集上述人群的粪便样品后采用试剂盒进行dna提取,得到提取的 dna样本。
46.2、宏基因组高通量测序及分析
47.采用illumina hiseq测序平台测序,共获得5,933,464.129,999,99mbp 的原始数据(raw data)(平均数据量7,756.16mbp),经过质控得到 5,885,567.3mbp的有效数据(clean data)(平均数据量为7,693.55mbp),经过单样品组装及混合组装后,共得到97,165,177,458bp的scaftigs。对各样品及混合组装的结果,采用metagenemark软件进行基因预测,共得到 123,459,411个开放阅读框(orfs)(平均为161,385),经过去冗余后,共获得6,727,989个orfs,总长为4,584.45mbp,其中完整基因的个数为 3,686,582,所占比例为54.79%。非冗余基因集与micronr库进行blastp比对,运用lca算法进行物种注释,注释到属和门的比例分别为65.11%, 86.00%。
48.(1)测序数据预处理
49.质控结果概述:总共测序数据量为5,933,464.129,999,99mbp,平均测序数据量为7,756.16mbp,质控后总体数据量及平均数据量分别为 5,885,567.3mbp,7,693.55mbp,质
控的有效数据率为99.19%。
50.数据预处理的具体处理步骤如下:
51.1)去除所含低质量碱基(质量值《=38)超过一定比例(默认设为40bp) 的reads;
52.2)去除n碱基达到一定比例的reads(默认设为10bp);
53.3)去除与adapter之间overlap超过一定阈值(默认设为15bp)的reads;
54.4)如果样品存在宿主污染,需与宿主数据库进行比对,过滤掉可能来源于宿主的reads;
55.(2)metagenome组装
56.组装结果概述:共组装得到105,500,331,957bp的scaffolds,平均长度为1,934.98bp,最大长度为1,733,071bp,n50为4,517.84bp,n90为 692.50bp;从n处打断scaffolds,生成scaftigs,共得到97,165,177,458bp 的scaftigs,scaftigs平均长度为1,868bp,n50为4,139bp,n90为678bp。
57.metagenome组装的具体处理步骤如下:
58.1)经过预处理后得到clean data,使用soap denovo组装软件进行组装;
59.2)对于单个样品,首先选取一个k-mer(默认选取55)进行组装,得到该样品的组装结果;
60.3)将组装得到的scaffolds从n连接处打断,得到不含n的序列片段,称为scaftigs(i.e.,continuous sequences within scaffolds);
61.4)将各样品质控后的cleandata采用bowtie2软件比对至各样品组装后的scaftigs上,获取未被利用上的pe reads;
62.5)将各样品未被利用上的reads放在一起,进行混合组装,组装时,考虑到计算消耗和时间消耗,只选取一个kmer进行组装(默认-k 55),其他组装参数与单样品组装参数相同;
63.6)将混合组装的scaffolds从n连接处打断,得到不含n的scaftigs 序列;
64.7)对于单样品和混合组装生成的scaftigs,过滤掉500bp以下的片段,并进行统计分析和后续基因预测;
65.(3)基因预测及丰度分析
66.基因预测结果概述:一共预测得到123,459,411条orfs,平均每个样品161,385条orfs;经去冗余后,得到6,727,989条orfs,去冗余后的orfs 总长为4,584.45mbp,平均长度681.4bp,gc含量为45.77%,其中,完整基因有3,686,582个,占所有非冗余基因总数的54.79%。
67.基因预测基本步骤:
68.1)从各样品及混合组装的scaftigs(》=500bp)出发,采用metagenemark 进行orf(open reading frame)预测及过滤;
69.2)对各样品及混合组装的orf预测结果,采用cd-hit软件进行去冗余;
70.3)将各样品的clean data比对至去冗余后的代表性基因上,计算得到基因在各样品中比对上的reads数目;
71.4)过滤掉在各个样品中,不存在支持reads数目》2的基因,获得最终用于后续分析的gene catalogue(unigenes);
72.5)从比对上的reads数目及基因长度出发,计算得到各基因在各样品中的丰度信息;
73.6)基于gene catalogue中各基因在各样品中的丰度信息,进行基本信息统计,core-pan基因分析,样品间相关性分析,及基因数目韦恩图分析。
74.(4)物种注释
75.物种注释结果概述:原始去冗余后的预测基因共有6,727,989条,其中,能够注释到nr数据库的orfs数目为5,317,849(79.04%),在能够注释到nr数据库的orfs中,注释到界水平的比例为88.82%,门水平的比例为86.00%,纲水平的比例为81.43%,目水平的比例为80.77%,科水平的比例为69.52%,属水平的比例为65.11%,种水平的比例为49.00%。其中占主导地位的门主要包括firmicutes,proteobacteria,bacteroidetes等。组间具有显著性差异的门主要有k__bacteria\;p__actinobacteria, k__bacteria\;p__chlamydiae,k__archaea\;p__euryarchaeota等。
76.注释基本步骤:
77.1)使用diamond软件将unigenes与从ncbi的nr(version:2018.01) 数据库中抽提出的细菌(bacteria)、真菌(fungi)、古菌(archaea)和病毒(viruses)序列进行比对(blastp,evalue《=1e-5);
78.2)比对结果过滤:对于每一条序列的比对结果,选取evalue《=最小 evalue*10的比对结果进行后续分析;
79.3)过滤后,采取lca算法(应用于megan软件的系统分类),将出现第一个分支前的分类级别,作为各序列的物种注释信息;
80.4)从lca注释结果及基因丰度表出发,获得各个样品在各个分类层级 (界门纲目科属种)上的丰度信息和基因数目信息;
81.5)从各个分类层级(界门纲目科属种)上的丰度表出发,进行krona 分析,相对丰度概况展示,丰度聚类热图展示,pca和nmds降维分析, anosim组间(内)差异分析,组间差异物种的metastat和lefse多元统计分析。
82.3、分类模型的构建
83.利用上述流程得到的微生物物种丰度信息表建立机器学习分类模型。
84.基于xgboost(extreme gradient boosting)选取不同数量的肠道微生物特征对上述大众人群和特定人群做分类,并使用十折交叉验证的方式最终取auc值(roc曲线下方的面积大小)的平均值,每次随机取数据的70%作为训练集,剩余的30%作为测试集,最终筛选出最优分类模型包含的23 个特征:s_actinobacillus succinogenes、s_actinomyces sp.hmsc035g02、 s_alistipes indistinctus、s_citrobacter sp.mgh110、s_clostridium sp.cag_58、 s_cohnella sp.ov330、s_fibrobacter sp.uwov1、s_klebsiella sp.obrc7、 s_klebsiella variicola、s_kluyvera ascorbata、s_kytococcus sedentarius、 s_lactobacillus kefiranofaciens、s_lactococcus garvieae、s_lysobacterenzymogenes、s_olsenella umbonata、s_paenibacillus massiliensis、s_pantoeaananatis、s_peptostreptococcus sp.d1、s_porphyromonas sp.hmsc065f10、 s_prevotella aurantiaca、s_slackia piriformis、s_candidatus saccharibacteriaoral taxon tm7x、s_lachnospiraceae bacterium khcpx20。
85.三、实验结果
86.基于23个特征构建的模型为最优模型。图1为每个特征的贡献值结果图;图2为特征数目与auc值对应关系图。
87.图3为最优模型的roc曲线,auc=0.97
±
0.02,p《0.01,说明利用这些微生物构建的模型可以准确区分大众人群和特定人群。
88.以上结合附图详细描述了本技术的优选实施方式,但是,本技术并不限于上述实施方式中的具体细节,在本技术的技术构思范围内,可以对本技术的技术方案进行多种简单变型,这些简单变型均属于本技术的保护范围。
89.另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本技术对各种可能的组合方式不再另行说明。
90.此外,本技术的各种不同的实施方式之间也可以进行任意组合,只要其不违背本技术的思想,其同样应当视为本技术所公开的内容。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
