由羟基化脂肪酸制备弹性体的方法和通过这样的方法获得的弹性体
1.本发明属于聚合物领域,并且更特别地为弹性体(即具有橡胶弹性特性的聚合物)领域。
2.更特别地,本发明涉及用于基于多羟基化脂肪酸或者这样的脂肪酸的酯来制备聚合物的方法,以及可通过这样的方法获得的聚合物。
3.ω-羟基化脂肪酸定义为在ω位包含至少一个羟基oh的脂肪酸,即该羟基oh由脂肪酸链的最后一个碳原子携带,链的第一个碳原子是分子的羧基的碳原子。按照惯例,表述ω-羟基化脂肪酸通常并且在本说明书中是指包括位于ω位的单个羟基的脂肪酸。此外,术语“多羟基化脂肪酸”也以常规方式表示包含多个羟基的脂肪酸,其中至少一个羟基位于ω位。因此,二羟基化脂肪酸包含两个羟基,其中至少一个位于ω位,三羟基化脂肪酸包含三个羟基,其中至少一个位于ω位,依此类推。
4.具有橡胶特性的聚合物或弹性体在许多领域获得应用,特别是在电子、复合材料、零件防腐蚀保护等领域,其中它们的弹性特性可有利地用于制造多种产品,适用于多种行业,如汽车行业、航空行业、医疗行业等。
5.此外,农业资源的价值化是当前绿色化学研究的挑战之一。化学工业的重要目标是实施可再生、可生物降解和无毒的原料用于制备多种物质和产品,并且特别是用于制备聚合物材料。
6.本发明符合该目的。更特别地,本发明人对植物来源的化合物感兴趣,该化合物是ω-羟基化和多羟基化类型的脂肪酸。这些具有长碳链的脂肪酸可以从植物中提取,特别是从水果和蔬菜的角质层中提取,并且更特别地是从进入这些角质层组合物中的角质中提取。虽然在自然界中大量存在,但它们目前价值不大。
7.本发明旨在提供在制备具有弹性变形能力的聚合物的特定背景下用于开发这样的羟基化脂肪酸的解决方案。
8.现有技术已经提出了这样的解决方案以用于形成生物聚酯,特别是通过由benitez等在biochimica and biophysica acta,2004,1674:1-3中的出版物进行了说明。该文献描述了在甲苯中,在存在催化剂十二烷基苯磺酸的情况下,通过缩聚反应由从番茄角质中提取的单体制备与天然角质相同的生物聚酯的方法。最近,benitez等在materials,2018,11,2211中的出版物提出了类似的方法,其中缩聚反应在不存在催化剂的情况下进行。
9.文献wo 2017/147708描述了使用催化剂由ω-羟基化脂肪酸和2-丁基-2-乙基-1,3-丙二醇制备聚合物的方法。
10.本发明人现已发现,可以在不使用催化剂的情况下,通过适当选择试剂和实施的操作模式由多羟基化脂肪酸或其酯制造具有类似橡胶的弹性特性的聚合物,该聚合物的特性,特别是弹性特性和热机械特性可以准确且容易地受控。
11.因此,本发明旨在提供由多羟基化脂肪酸或者由多羟基化脂肪酸的酯制备其橡胶特性可以以受控方式调节的弹性体的方法。
12.本发明的另外目的是该方法易于实施,其不使用对人和动物有毒和/或对环境有害的任何产品,并且其允许形成可回收的弹性体,而且成本低。
13.因此,根据第一方面,本发明提供了用于基于至少一种多羟基化脂肪酸来制备聚酯型聚合物,更特别地为三维热固性网络形式的弹性体的方法。该方法包括以下连续步骤:
[0014]-通过混合/汇集以下来制备反应介质:
[0015]-单体,其选自多羟基化脂肪酸,以及多羟基化脂肪酸与具有直链或支链、饱和或不饱和、任选经取代的脂肪链的醇的酯,所述脂肪链包含1至18个碳原子;或者羟基化脂肪酸和/或多羟基化脂肪酸与具有直链或支链、饱和或不饱和、任选经取代的脂肪链的醇的酯的混合物,所述脂肪链包含1至18个碳原子,相对于羟基化脂肪酸和/或酯的所述混合物的总重量,所述混合物包含按重量计至少20%的所述单体,
[0016]-以及不同于所述单体并且在适当情况下不同于所述混合物中的所述羟基化脂肪酸和/或所述酯(羟基化脂肪酸与具有直链或支链、饱和或不饱和、任选经取代的脂肪链的醇的酯,所述脂肪链包含1至18个碳原子)的多元醇,
[0017]
该反应介质不含催化剂,
[0018]-以及加热所述反应介质以进行所述单体与所述多元醇的共聚和由此形成的聚合物的交联的步骤。该加热步骤的至少初始阶段在减压下进行。由此,应当理解的是在加热步骤的早期阶段,在该步骤的至少开始时施加减压。
[0019]
通过这样的方法从由多羟基化脂肪酸或者这样的脂肪酸的酯组成的所述单体形成的聚合物有利地不仅包含涉及在单体ω位的羟基的直链/伯酯键,而且还包含涉及单体的碳链中包含的其它羟基的仲酯键。这导致在所获得聚合物的结构中具有多个分支的交联模式,这是可调节的,允许控制聚合物的特性,并且比现有技术提出的用具有单羟基官能团的ω-羟基化脂肪酸获得的要有利得多。
[0020]
优选地,所有这些步骤在单个容器中进行,优选地在具有适合于在完成该方法时获得的聚合物的预期应用的形状的模具中进行。优选地,加热步骤的所有阶段还通过相同的加热装置进行。
[0021]
在其中实施根据本发明方法的加热装置本身是常规的。特别地,其由炉子组成,所述炉子可具有一个或数个温度区域、配备有用于在其中建立减压的装置,以及优选地用于搅拌放置在其中的容器/模具的装置。
[0022]
除了不含催化剂,即反应促进剂/促进剂之外,反应介质还优选地不含溶剂,并且优选地也不含链阻断剂。
[0023]
有利地,根据本发明的方法的加热步骤在允许通过单体的酸官能团实现多元醇的一个或更多个羟基基团的酯化的条件下实施,从而能够使聚合物交联以形成热固性三维网络。然后获得固体形式的弹性体。根据所使用的特定单体和多元醇以及弹性体所期望的交联度,确定为获得这种三维热固性网络而应用的时间和温度条件在本领域技术人员的技能范围内。有利地,该弹性体的弹性和热机械特性可以根据反应介质中多元醇的初始含量进行调节。
[0024]
此外,在加热步骤的至少初始阶段期间施加减压确保了在聚合反应期间产生的水在形成时消除,这有利地允许将该反应的热力学平衡移向酯的形成并提高反应动力学。本发明人还注意到,出人意料地,在加热步骤的初始阶段期间施加的压力水平影响了反应期
间酯化的单体和多元醇的羟基官能团的数量和性质,并且因此影响了在完成根据本发明的方法时形成的弹性体的橡胶弹性特性。通过设置在加热步骤的初始阶段期间施加的压力,因此可以以受控的方式调节这些特性。
[0025]
在本发明的一些优选实施方案中,加热步骤的至少初始阶段在0至900mbar的压力下进行。
[0026]
优选地,在加热步骤的至少初始阶段期间施加的压力为400至800mbar。本发明人出人意料地发现,通过施加该数值范围内的压力获得的聚合物的热机械特性,特别是储能模量(动态机械分析(dvnamic mechanical analysis,dma)中材料的弹性响应)和表观交联密度都特别稳定。因此,通过特别是能够控制聚合物内的酯化多元醇的含量,这样的特征允许非常良好地控制聚合物的结构和网孔尺寸,以及其结晶度。在加热步骤的初始阶段期间施加400至800mbar范围内的压力不仅允许提高聚合反应的速率,而且可以使未交联多元醇的量最小化。
[0027]
优选地,加热步骤的初始阶段的持续时间基本上对应于在所实施的操作条件下达到在反应介质中形成的聚合物的凝胶点所需的时间。特别地,对于给定的单体浓度,该持续时间取决于加热温度和反应介质中多元醇的浓度。
[0028]
因此,以常规方式,对于导致形成化学交联聚合物网络的任何方法,凝胶点在本文中定义为固体三维网络出现在液体反应介质中的时刻。凝胶时间对应于反应开始与反应介质中达到凝胶点的时刻之间的时间间隔。这种固体网络的出现导致流动黏度的发散,其在凝胶时间趋于无穷大。此外,这种固体网络本质上不溶于溶剂。因此,可以通过在能够溶解初始反应介质组分的溶剂中的溶解度测试来检测其出现,即凝胶点的出现。
[0029]
对于根据本发明制备聚合物的方法的每组特定操作条件,确定达到该凝胶点所需的凝胶时间在本领域技术人员的技能范围内。为此,本领域技术人员尤其可以凭经验进行,例如通过反应介质在不同反应时间之后,在能够溶解初始反应介质的溶剂中的溶解测试。然后凝胶时间将为导致观察到不溶于用于溶解度测试的溶剂中的部分的最短反应时间与反应介质在存在该溶剂的情况下保持完全可溶的最长反应时间之间。例如,在不同的反应时间之后,10mg反应介质的样品因此可以掺入1ml溶剂中,例如乙醇中。在涡旋5分钟之后,观察到混合物中可能出现不溶性部分,这表明已经达到凝胶点并且超过了凝胶时间的事实。这样的溶解度测试可以以非常接近的间隔进行,例如每10秒,以便尽可能准确地确定凝胶时间。该溶解度测试可以在对照反应介质上进行,特别是在小体积的反应介质上,以确定聚合物在随后将用于实施根据本发明的方法本身的操作条件下的凝胶时间。
[0030]
优选地,初始阶段的持续时间为30分钟至5小时,优选为1小时至3小时。
[0031]
根据本发明的方法可以同样实施选自以下的单一单体或者多个这样的单体:多羟基化脂肪酸和多羟基化脂肪酸与具有直链或支链、饱和或不饱和、任选经取代的脂肪链的醇的酯,所述脂肪链包含1至18个碳原子。
[0032]
此外,在本说明书中,术语“多元醇”是指单一多元醇和多元醇的混合物两者。
[0033]
优选地,反应介质包含按重量计小于10%的除多元醇和选自以下的单体之外的组分:多羟基化脂肪酸和多羟基化脂肪酸与具有直链或支链、饱和或不饱和、任选经取代的脂肪链的醇的酯,所述脂肪链包含1至18个碳原子。因此,多元醇和单体的混合优选地占反应介质的按重量计至少90%。
[0034]
根据本发明的方法可进一步满足单独或以其技术上可行的组合中的任一种实施的以下描述的一个或更多个特征。
[0035]
实施的多元醇可包含一个或数个酸官能团,或者其可以不包含任何酸官能团。优选地,其不是羟基化脂肪酸,并且尤其不是多羟基化脂肪酸或ω-羟基化脂肪酸。
[0036]
在本发明一些特别优选的实施方案中,多元醇是三醇或包含三醇。优选地,其由甘油组成,所述甘油可以单独使用或与一种或数种其他多元醇,特别是一种或数种其他三醇混合使用。
[0037]
特别地,甘油具有基于生物的来源和对生物无毒性的优点。此外,甘油是生物燃料生产的主要废物,因此根据本发明的方法然后可以成为从农业和工业废物回收的一部分。
[0038]
在本发明的一些具体实施方案中,相对于反应介质的总重量,反应介质包含浓度为按重量计1至25%的多元醇。有利地,这样的浓度范围允许在不超过约50小时的时间内以良好的反应动力学获得具有橡胶弹性特性的聚合物,该橡胶弹性特性的程度可根据反应介质中多元醇的精确浓度进行调节,从而允许以可能对于预期应用来说最合适的方式来精细调节这些特性。
[0039]
在该浓度范围内,聚合物中存在的按重量计多于85%,并且甚至在大多数构造中按重量计多于90%的多元醇参与形成的聚合物的交联网络。这应该被理解为意指聚合物中存在的按重量计多于85%,并且最经常按重量计多于90%的多元醇通过至少一个其羟基官能团在聚合物内参与酯键。不参与聚合物内共价键的其他多元醇分子对后者具有增塑剂的作用。
[0040]
在本发明的一些具体实施方案中,相对于反应介质的总重量,反应介质包含按重量计15至25%的多元醇。本发明人已经出人意料地发现,多元醇的这样的浓度范围允许形成具有结晶区的聚合物,随初始多元醇浓度本身高,聚合物的结晶状态越来越高。这样的结晶区的存在使得通过根据本发明的方法形成的聚合物对于许多应用来说特别有意义。
[0041]
此外,非常有利地,在50℃下测量的由根据本发明方法形成的聚合物的储能模量(在动态机械分析(dma)中的弹性响应)在多元醇为按重量计15至25%范围内几乎无变化。然后,该储能模量对应于材料低变形时的弹性模量。在50℃的温度下,这些材料表现得像具有在兆帕范围内的弹性模量的橡胶。
[0042]
flory橡胶弹性理论的应用然后允许估计弹性体网络的表观交联密度,其与弹性模量成正比并因此在初始反应介质中多元醇为按重量计15至25%范围内也有利地几乎恒定。
[0043]
在根据本发明的方法中实施的多羟基化脂肪酸可以是直链或支链的,即由具有直链或支链脂肪链的羧酸组成,该脂肪链带有数个羟基官能团,其中至少一个位于所述链的末端位置。根据本发明实施的多羟基化脂肪酸可特别对应于通式(i):
[0044]
(ho)c
nh2n-m-2p
(oh)mcooh
ꢀꢀ(i)[0045]
其中:
[0046]
n为7至21、优选12至20、更优选13至19、优先地15至17的整数,
[0047]
m为大于0的整数,优选1至3、并且优先地等于1,
[0048]
p表示在所述脂肪酸中包含的不饱和数目,并且为0至3的整数,优选等于0。
[0049]
特别地,可根据本发明实施的多羟基化脂肪酸酯可对应于通式(ii):
[0050]
(ho)c
nh2n-m-2p
(oh)mcoor
ꢀꢀ
(ii)
[0051]
其中:
[0052]
n、m和p如前定义,
[0053]
并且r表示直链或支链、饱和或不饱和、任选经取代的脂肪链,该脂肪链包含1至18个碳原子,优选1至6个碳原子,r优先地为甲基或乙基。
[0054]
在本说明书中,“脂肪链”应理解为非芳香族开碳链。
[0055]
优选地,单体是多羟基化脂肪酸的甲酯或乙酯。
[0056]
可以在根据本发明的方法中实施的多羟基化脂肪酸的一些优选特征在下文中详细公开。相同的特征可以相同地转置为多羟基化脂肪酸与具有直链或支链、饱和或不饱和、任选经取代的脂肪链的醇的酯,所述脂肪链包含1至18个碳原子。
[0057]
优选地,在根据本发明的方法中作为单体实施的多羟基化脂肪酸是二羟基化脂肪酸,即其碳链带有两个羟基,其中一个位于链末端。优选地,多羟基化脂肪酸带有单一酸官能团。
[0058]
因此,在本发明的一些具体实施方案中,在该方法中实施的单体是二羟基化脂肪酸,优选具有单一酸官能团。
[0059]
可在根据本发明的方法中作为单体实施的多羟基化脂肪酸的实例包括但不限于10,16-二羟基十六烷酸或9,10,18-三羟基十八烷酸。
[0060]
优选地,其由下式(i’)的10,16-二羟基十六烷酸组成:
[0061][0062]
特别适用于实施根据本发明的方法的酯包括10,16-二羟基十六烷酸的甲酯和乙酯。
[0063]
在根据本发明的方法中作为单体实施的多羟基化脂肪酸可以是化学合成的。另外地,它们可通过酶促方法或者通过酸或碱水解从植物中提取,更特别地从植物的角质层中提取,并且更具体地从角质中提取。
[0064]
角质是通过酯键交联的多羟基化脂肪酸(主要是c16和c18)的聚合物网络,其与高等植物叶子和果实的防水有关。其是植物角质层的主要组分,所述角质层是覆盖植物叶子和果实地上部分的连续的细胞外脂质膜。
[0065]
优选地,在根据本发明的方法中用作单体的多羟基化脂肪酸是从番茄中提取的,这种多羟基化脂肪酸的优点在于从一个物种到另一个物种的角质组成均一性大,该角质还具有以在很大程度上占主导地位的量存在的组成单体:10,16-二羟基十六烷酸,其以按重量计多于80%存在于其中。
[0066]
番茄转化废物称为番茄渣,其包含大量的角质,更具体地为60%至70%的角质。据估计,全世界每年生产4至5百万吨番茄渣。因此,根据本发明的方法可以非常有利地允许回收由番茄渣形成的农业和工业废物,这从环境角度和经济角度二者来看都是有利的。
[0067]
在本发明的一些具体实施方案中,根据本发明的方法中使用的单体通过使角质,
优选番茄角质解聚而获得。
[0068]
另外地,在根据本发明的方法中作为单体实施的多羟基化脂肪酸可以从其他植物中提取,例如苹果(malus pumila)、酸橙(citrus aurantium)、蚕豆(viciafaba)、野樱桃(prunus avium)、蔓越莓(vaccinium macrocarpon)、葡萄柚(vitis vinifera)、豌豆籽(pisum sativum)、醋栗(ribes grossularia)、木瓜(malabar papaiarnarum)、龙舌兰叶(agave americana)、葡萄柚籽(citrus paradisi)、柠檬(citrus limon)、酸橙(citrus aurantifolia)、番木瓜(carica papaya)、洋葱(allium cepa)、越橘(vaccinium vitis idaea)、咖啡叶(rubiaceaecoffea)、玫瑰果(rosa canina)、南瓜(cucurbita pepo)等。
[0069]
根据本发明可以实施任何从植物中提取角质的过程,特别是用于破碎番茄渣或番茄外皮的过程,以及任何用于使该角质解聚的过程,以获得方法中用作单体的多羟基化脂肪酸。基本上,这些方法包括将番茄渣或其他植物元素(例如苹果)破碎以提取角质,然后将角质水解以获得其组成单体。在分离之后,通过物理分离方法或通过液-液萃取,角质因此被化学水解,特别是通过在有机介质中的碱性途径,或使用特定酶,特别是角质酶。
[0070]
可以为此目的实施的方法的一个实例在文件wo 2015/028299中进行了描述。该方法图示地包括番茄外皮的热处理,然后将其引入到碱性溶液中,例如浓度为0.5m至6m,温度为20℃至130℃(例如65℃至130℃)的氢氧化钾溶液。然后,对溶液进行过滤并随后酸化,特别是用浓度为12m至6m的盐酸酸化。在例如以10000至14000rpm离心15至20分钟之后,将沉淀物例如用脱矿质水进行洗涤,然后在适当的情况下干燥。
[0071]
根据本发明的一个优选方法在于:在倾析番茄渣以回收外皮以及将由此回收的外皮干燥、研磨和脱脂的步骤之后,将其在碱性介质中在醇溶剂中水解。例如,这样的水解可以通过将脱脂和脱水的外皮在50℃下浸入醇氢氧化钾(例如由5%氢氧化钾在无水乙醇中形成)中6小时5天,优选2天来进行。之后,可以在真空下过滤混合物,并通过旋转蒸发器除去乙醇。可将所得组合物中包含的脂肪酸沉淀,特别是在ph为2至3的水中,例如在37%的盐酸溶液中,然后回收,特别是通过离心,例如以9000rpm离心20分钟。在将获得的沉淀物用脱矿质水清洗并冷冻干燥之后,以60至70%的产率获得油性组合物,该组合物基本上包含脂肪酸,主要是10,16-二羟基十六烷酸。
[0072]
更具体地,在角质解聚操作完成之后获得的这种油性组合物包含按重量计至少85%的脂肪酸。10,16-二羟基十六烷酸优选地在其中占这些脂肪酸的按重量计至少88%。还包含在380、288和225nm处吸收的酚类和类胡萝卜素化合物以及少量的其他物质(特别是浓度为按重量计约0.5至3%的酚类化合物)的该组合物可有利地直接用于根据本发明的方法。
[0073]
根据本发明可实施的多羟基化脂肪酸酯可根据对于本领域技术人员来说本身常规的任何方法通过相应脂肪酸的酯化来制备而无需催化剂或优选用酸催化剂。它可以另外通过包含在植物角质中的多羟基化脂肪酸的酯交换来获得,所述酯交换优选地在酸催化剂或碱性催化剂下,使用强碱或醇化物例如甲醇钠或乙醇钠进行操作。用于进行脂肪酸的酯化或酯交换反应的醇优选地包含1至18个碳原子,并且优选1至8个碳原子。该醇优选地选自甲醇、乙醇、丙醇、丁醇、戊醇、己醇及其异构体包括2-乙基丁醇、庚醇及其异构体例如2-庚醇、辛醇及其异构体例如2-乙基己醇,以及异丙醇、2-甲基-丙醇、2-甲基-丙-2-醇、丁-2-醇、戊醇、2-甲基-丁醇、3-甲基-丁醇、2,2-二甲基丙醇、戊-3-醇、戊-2-醇、3-甲基丁-2-醇、
2-甲基丁-2-醇、格尔伯特醇例如2-丙基-庚醇、2-丁基-辛醇。
[0074]
例如,可在根据本发明的方法中实施的多羟基化脂肪酸酯可以通过在50至70℃的温度下使番茄外皮在存在酸(例如2至5%的浓硫酸)的情况下与醇,特别是甲醇或乙醇接触至少6小时而直接从这些外皮生产。酯交换反应完成之后获得的酯可以通过向反应介质添加水然后离心来纯化。
[0075]
在本发明的一些具体实施方案中,反应介质的制备包括将多元醇与由角质、优选番茄角质或苹果角质的解聚产生的组合物混合。相对于组合物中包含的羟基化脂肪酸的总重量,优选富含二羟基化脂肪酸的这种组合物优选地包含按重量计至少20%,优选按重量计至少80%的单体。根据已用于进行角质解聚的操作模式,单体可以以酸的形式或以酯的形式存在于其中。
[0076]
在本发明的一些具体实施方案中,由角质解聚产生的这种组合物相对于组合物总重量优选地包含按重量计至少20%,优选至少80%的单体。
[0077]
优选地,其中包含的单体是二羟基化脂肪酸。
[0078]
优选地,反应介质基本上仅由这样的组合物和多元醇组成。
[0079]
优选地,该反应介质及其允许形成的弹性体完全是基于生物的。
[0080]
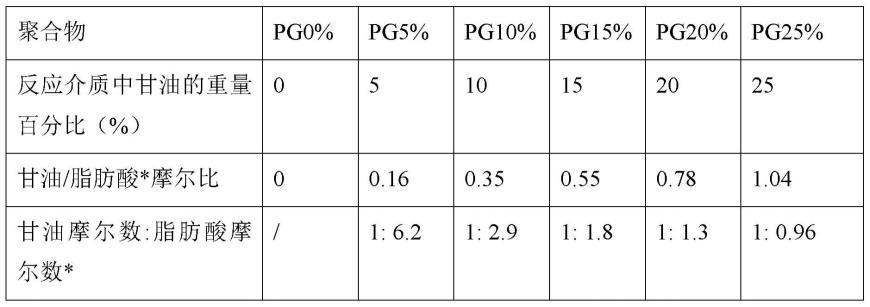
在本发明的一些具体实施方案中,多元醇(特别是甘油)与由角质在反应介质中聚合产生的单体或组合物之间的摩尔比为0.16至1.04。当单体包含在由角质解聚产生的组合物中时,该比值适用于多元醇(特别是甘油)与该组合物之间,然后通过将其摩尔质量同化为10,16-二羟基十六烷酸的摩尔质量来确定其摩尔数。
[0081]
优选地,根据本发明的方法的反应介质的制备步骤在高于或等于50℃的温度下进行。这样的特征有利地促进了单体与多元醇的均匀混合,并且当反应介质不含任何溶剂时更是如此。
[0082]
优选地,选择在加热步骤期间实施的操作条件以不引起反应介质中存在的多元醇的蒸发。
[0083]
在本发明的一些具体实施方案中,加热步骤在120至200℃的温度范围,特别是在约150℃的温度下进行。在加热步骤的初始阶段(优选地定义为在达到聚合物的凝胶点之前发生的阶段)期间施加的温度与在加热步骤的第二阶段期间施加的温度相比可以相同或不同,后者被定义为在初始阶段之后发生的直到加热步骤结束的阶段。
[0084]
优选地,加热步骤进行至少4小时,优选4至60小时并且优选5至28小时例如22至28小时的时间。
[0085]
特别地,加热步骤的初始阶段可以进行优选为30分钟至5小时,优选为1至3小时的时间,特别是约90分钟的时间。
[0086]
如前所述,在本发明的一些特别有利的优选实施方案中,加热步骤的初始阶段在0至900mbar、优选100至900mbar、并且优先地400至800mbar的压力下进行。这样的特征不仅促进了从反应介质中形成的水的蒸发并因此促进了多元醇被ω-羟基化脂肪酸酯化的反应动力学,而且出乎意料的是,还使得酯化的单体和多元醇(即参与形成的聚合物内的酯键)的重量百分比以及该聚合物的弹性特性最大化。特别地,低于100mbar的压力的实施导致多元醇,特别是甘油从反应介质中蒸发,并导致形成的聚合物的橡胶弹性特性不如施加的压力高于或等于200mbar时令人满意;并且高于900mbar的压力的实施降低了多元醇和存在的
单体的酯化速率。在0至900mbar的范围内,通常观察到施加的压力越低,通过根据本发明的方法形成的聚合物中游离羟基官能团的比值越低,这可以通过红外光谱(通过观察在3500cm-1处对应于游离羟基的带)分析聚合物来容易地检查。此外,关于这些游离羟基官能团的性质,当施加的减压在本发明建议的优选值范围内时,在单体是二羟基化脂肪酸的情况下,在其中羟基官能团全部酯化的单体片段在聚合物中的相对比例不超过80%,并且优选70%。本发明人v发现,在其中羟基官能团全部酯化的脂肪酸片段在聚合物中的相对比例越高,后者的橡胶特性越不太好。此外,其中游离仲羟基官能团保留的单体片段的相对比例小于或等于40%,这也证明对于聚合物的弹性特性是有利的。
[0087]
优选地,根据本发明的方法包括,在加热步骤期间,在达到所述聚合物的凝胶点之前,消除反应介质中存在的气泡的步骤。该步骤可以通过任何方式进行,例如通过手动或自动搅拌反应介质进行。它可以连续进行,特别是在加热步骤的初始阶段的整个持续时间内,或者在加热步骤期间的一个或更多个时刻偶尔进行,并且在这种情况下,优选地在刚好达到聚合物的凝胶点之前,例如在达到聚合物的凝胶点1至5分钟之前进行至少一次。在本发明的一个具体实施方案中,消除反应介质中存在的气泡的步骤包括至少一个以下的阶段:在反应介质中存在的聚合物刚好达到其凝胶点之前(例如,当达到上述溶解度测试中反应介质在存在溶剂的情况下保持完全可溶的最长反应时间时)对反应介质进行搅拌。
[0088]
在偶尔搅拌反应介质以去除已在其中形成的气泡的阶段期间可以任选地中断反应介质的加热。
[0089]
加热步骤的第二阶段可以进行足够长的时间以获得期望的弹性体交联度。该持续时间优选为至少4小时,并且优选为4至48小时,例如约24小时。
[0090]
优选地,在加热步骤的第二阶段之前或期间不向反应介质中添加试剂或其他物质。
[0091]
优选地,选择在加热步骤期间实施的操作条件以使聚合物的交联度高于或等于50%,特别是50至90%。
[0092]
根据本发明的方法有利地允许形成这样的弹性体,其特性(特别是弹性特性)和玻璃化转变温度可以调节,特别是根据所实施的多元醇(优选甘油)的含量,在该聚合物中甘油特别是既作为聚合物的组成单体又作为增塑剂,以及根据加热步骤期间施加的减压值和施加该减压的持续时间。
[0093]
根据第二方面,本发明涉及可通过根据本发明的方法获得的聚酯型疏水聚合物。
[0094]
该聚合物的单体单元是多羟基化脂肪酸。
[0095]
特别地,该聚合物可以由至少一种多元醇和至少一种多羟基化脂肪酸形成,或由如上文所定义的这样的脂肪酸的酯形成,特别是基于甘油和10,16-二羟基十六烷酸,并且在适当的情况下基于其他单体。
[0096]
其橡胶弹性根据包含在其中的多元醇含量,特别是甘油含量而变化。这种可定性为弹性体的聚合物特别是更具弹性,因为它包含更多的多元醇,特别是甘油。
[0097]
其玻璃化转变温度可在-12.5至-25℃变化,这取决于其多元醇,特别是甘油的含量。该玻璃化转变温度通过差示焓分析(differential enthalpyanalysis,dea或差示扫描量热法(differential scanning calorimetry,dsc))测量,包括对聚合物样品进行从-50℃温度到80℃温度的温度扫描,升温斜率为3℃/分钟。该分析优选对置于密封铝胶囊中的
10mg量的聚合物进行。
[0098]
根据本发明的聚合物具有非常高的分子量和50至90%的交联率。该交联率定义为参与聚合物内酯键的单体的羟基官能团的比例,可以以其本身常规的任何方式来确定,特别是通过标记聚合物的游离羟基官能团来确定。换言之,根据本发明的聚合物使得由单体单元携带的游离的羟基官能团的比例为10至50%。对于本领域技术人员而言,该游离羟基官能团比例可以以其本身常规的任何方式来确定。例如,其可以通过聚合物内游离羟基官能团的化学标记来确定,这种标记对聚合物的解聚反应具有抗性,例如通过苄基醚化,如philippe等,2016,plant physiology,doi:10.1104/pp.15.01620)的出版物中所述;然后解聚并测量由此释放的单体中标记的羟基官能团(其因此不参与聚合物中的共价键)和未标记的羟基官能团(其因此参与聚合物中的共价键)的各自比例。
[0099]
这种聚合物不溶于大多数溶剂,特别是水、乙醇、甲醇、异丙醇、氯仿、二氯甲烷、二甲亚砜和四氢呋喃,或它们的任何混合物。在这方面其与树状聚合物不同,后者可溶于许多溶剂。在本说明书中,“不溶”应理解为在室温下搅拌18小时后,未溶解的聚合物在其所浸入的溶剂体积中的重量百分比高于85%的事实。
[0100]
这种聚合物的结构的一个实例示于图1中,对于这种情况,其中单体为10,16-二羟基十六烷酸且多元醇为甘油。该聚合物包含其中羟基官能团均被酯化的二羟基化脂肪酸片段、其中保留游离伯羟基官能团的片段和其中保留游离仲羟基官能团的片段。
[0101]
聚合物中这些片段中的每一个的相对比例可以以其本身常规的任何方式确定。例如,可以应用philippe等在plant physiology,2016,170,807-820中的出版物中描述的方法。简而言之,该方法在于标记聚合物的游离羟基官能团,进行其解聚,分离获得的单体片段以及对其进行分析以评估标记的羟基官能团的存在和类型。
[0102]
作为可以应用的实验方案的一个实例,将5mg聚合物与50mg 2-苄氧基-1-甲基吡啶三氟甲磺酸盐和6mg氧化镁在1ml三氟甲苯中于90℃下在塞紧的玻璃管中混合24小时。然后,聚合物用二氯甲烷清洗并干燥。然后,使用弱碱(例如0.5m甲醇钠)在无水甲醇中使标记的聚合物解聚。这种解聚可以在60℃下进行16小时。在硅烷化之后,对该级分的等分试样进行取样并进样以通过气相色谱与质谱联用(gc-ms)进行分析。
[0103]
优选地,其中羟基官能团均被酯化的脂肪酸片段在聚合物中的相对比例不超过80%,并且优选不超过70%。本发明人已发现,其中羟基官能团均被酯化的脂肪酸片段在聚合物中的相对比例越高,聚合物的橡胶特性越差。高于80%的相对比例指示聚合物内存在显著量的支链,与橡胶特性不足有关,并且特别是与聚合物非常低的拉伸能力有关。
[0104]
优选地,根据本发明的聚合物来源于原材料回收,并且其本身是可回收的。
[0105]
这种聚合物由于其特性的模块化而可用于许多不同的应用。
[0106]
在本发明的一些具体实施方案中,聚合物具有以下特征中的至少一种,优选数种:
[0107]-其具有结晶区,
[0108]-其具有高于或等于40℃,更特别地为40℃至42℃的解链温度,
[0109]-其在50℃下的储能模量(动态力学分析中的弹性响应)为1.4至1.6mpa,
[0110]-其机械弛豫温度低于2℃,
[0111]-其杨氏模量为1.10至1.20mpa,
[0112]-其抗张强度为1.2至1.6mpa。
[0113]
所述聚合物还具有大于100%,并且甚至大于150%的断裂伸长率。
[0114]
鉴于下文中实施方式的实例,参照图1至图25,本发明的特征和优点将更清楚地显现,所述实施方式的实例仅为举例说明本发明而不是限制而提供,其中:
[0115]
图1示出了根据本发明的聚合物的结构的一个实例,该聚合物由甘油和10,16-二羟基十六烷酸获得。
[0116]
图2示出了显示以下的图表:在根据本发明用于由甘油和从番茄角质中提取的油性组合物制备聚合物的方法的实施完成之后,作为反应介质中初始甘油重量含量的函数的相对于反应介质中初始甘油重量的酯化甘油重量百分比。
[0117]
图3示出了显示以下的图表:在根据本发明用于由甘油和从番茄角质中提取的油性组合物制备聚合物的方法的实施完成之后,作为反应介质中初始甘油重量含量的函数的相对于反应介质中脂肪酸初始重量的酯化脂肪酸重量百分比。
[0118]
图4示出了分别对于以下通过傅里叶变换红外光谱获得的光谱:通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在初始重量含量为25%甘油,在加热步骤的初始阶段期间施加了400mbar的压力的情况下制备的聚合物(“pg25%”);以及从番茄角质中提取的这种油性组合物(“ch”)。
[0119]
图5示出了图4的光谱区域的放大图。
[0120]
图6示出了对于以下聚合物通过傅里叶变换红外光谱获得的光谱区域的放大图:该聚合物通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在初始甘油重量含量为15%,在加热步骤的初始阶段期间分别施加了0、200、400、600、800和1000mbar的压力的情况下制备。
[0121]
图7示出了显示以下的条形图:对于不含甘油的比较聚合物(“pg0%”),以及对于通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在甘油的初始重量含量分别为5至25%,在加热步骤的初始阶段期间施加了400mbar压力的情况下制备的聚合物(“pg5%”至“pg25%”),完全酯化(iiia)、仅在伯羟基官能团上酯化(iiic)和仅在仲羟基官能团上酯化(iiib)的来源于10,16-二羟基十六烷酸的片段在聚合物中的相对比例。
[0122]
图8示出了显示以下的条形图:对于通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在初始甘油重量含量为15%,在加热步骤的初始阶段期间分别施加了0、200、400、600、800和1000mbar压力的情况下制备的聚合物,完全酯化(iiia)、仅在伯羟基官能团上酯化(iiic)和仅在仲羟基官能团上酯化(iiib)的由10,16-二羟基十六烷酸产生的片段在聚合物中的相对比例。
[0123]
图9示出了显示以下的条形图:对于通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在甘油的初始重量含量分别为5至25%,在加热步骤的初始阶段期间施加了400mbar压力的情况下制备的聚合物(“pg5%”至“pg25%”),完全酯化(iva)、仅在1位或3位羟基官能团上酯化(ivb)和仅在2位羟基官能团上酯化(ivc)的由甘油产生的片段在聚合物中的相对比例。
[0124]
图10示出了显示作为反应介质中初始甘油重量含量的函数的以下聚合物的通过dsc测量的玻璃化转变温度的图表:该聚合物通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在加热步骤的初始阶段期间施加了400mbar压力下制备。
[0125]
图11示出了显示作为反应介质中初始甘油重量含量的函数的以下聚合物的通过
dma测量的机械弛豫温度的图表:该聚合物通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在加热步骤的初始阶段期间施加了400mbar压力下制备。
[0126]
图12示出了用于旨在确定根据本发明聚合物的机械特性的拉伸试验的受试试样的形状和尺寸。
[0127]
图13示出了对于由根据本发明聚合物形成的受试试样测量的作为反应介质中初始甘油重量含量的函数的杨氏模量,在加热步骤的初始阶段期间施加了400mbar的压力。
[0128]
图14示出了对于由根据本发明聚合物形成的受试试样测量的作为反应介质中初始甘油重量含量的函数的最大抗张强度,在加热步骤的初始阶段期间施加了400mbar的压力。
[0129]
图15示出了对于由根据本发明聚合物形成的受试试样测量的作为反应介质中初始甘油重量含量的函数的断裂伸长率百分比,在加热步骤的初始阶段期间施加了400mbar的压力。
[0130]
图16示出了由从番茄角质中提取的油性组合物在加热步骤的初始阶段期间施加了400mbar压力下制备的聚合物的通过dsc获得的曲线。
[0131]
图17示出了对于以下聚合物通过dsc获得的曲线:该聚合物通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在初始甘油重量含量为15%,在加热步骤的初始阶段期间施加了400mbar压力的情况下制备。
[0132]
图18示出了对于以下聚合物通过dsc获得的曲线:该聚合物通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在初始甘油重量含量为25%,在加热步骤的初始阶段期间施加了400mbar压力的情况下制备。
[0133]
图19示出了以下聚合物的xrd谱:该聚合物由从番茄角质中提取的油性组合物在加热步骤的初始阶段期间施加了400mbar压力下制备。
[0134]
图20示出了以下聚合物的xrd谱:该聚合物通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在初始甘油重量含量为25%,在加热步骤的初始阶段期间施加了400mbar压力的情况下制备。
[0135]
图21示出了对于以下聚合物获得的xrd光谱:该聚合物通过在加热步骤期间施加根据本发明的400mbar压力或施加1000mbar压力下,由从番茄角质中提取的油性组合物和由反应介质中重量含量为15%的甘油制备。
[0136]
图22示出了对于以下获得的xrd光谱:从番茄角质中提取的油性组合物(在a/中);以及根据本发明由该油性组合物和重量含量为20%的甘油在加热步骤期间施加了400mbar的压力下获得的聚合物(在b/中)。
[0137]
图23示出了显示以下的图表:一方面作为反应介质中初始甘油重量含量的函数的聚合物在50℃下的储能模量,以及另一方面作为反应介质中初始甘油重量含量的函数的聚合物表观交联密度,该聚合物通过根据本发明的方法由甘油和从番茄角质中提取的油性组合物在加热步骤的初始阶段期间施加了400mbar的压力下制备。
[0138]
图24示出了显示作为压力的函数的“储能模量/大气压下的储能模量”比值(e
′
/eatm比值)的曲线图,该压力是在根据本发明实施甘油和从番茄角质中提取的油性组合物,初始甘油重量含量为25%的方法的加热步骤期间施加的。
[0139]
图25示出了分别对于以下通过傅里叶变换红外光谱获得的光谱:通过根据本发明
的方法由甘油和从苹果角质中提取的油性组合物在初始重量含量为10%甘油,在加热步骤的初始阶段期间施加了400mbar压力的情况下制备的聚合物(“ppg10%”);以及从苹果角质中提取的这种油性组合物(“chp”)。
[0140]
实施例1-番茄角质
[0141]
由番茄渣制备包含ω-羟基化脂肪酸的组合物
[0142]
如下实施用于由甘油和从番茄角质中提取的油性组合物制备根据本发明生物弹性体的方法。
[0143]
通过倾析从番茄渣中分离番茄外皮。将这些外皮干燥、压碎并在索氏提取器中使用丙酮∶乙醇(1∶1)混合物通过回流脱脂2天。然后,将它们脱水。
[0144]
将200g如此预先脱蜡和脱水的番茄外皮悬浮在1l的在无水乙醇中制备的5%氢氧化钾koh溶液中。将混合物在50℃下加热16小时。然后,将悬浮液通过a0(160至250μm)大小的筛板在真空下过滤,通过蒸发减少乙醇的体积,然后将滤液用水稀释并使用37%盐酸hcl溶液酸化至ph 3至4。将由此形成的悬浮液在20℃下以8000rpm离心15分钟,然后回收离心沉淀,用水洗涤,然后真空干燥。由此获得了150g主要由脂肪酸(按重量计超过85%)以及少量未鉴定的色素(包括在300、288和225nm处吸收的着色物质)组成的油性组合物。通过气相色谱与质谱联用gc-ms/fid对该组合物的分析表明,该油性组合物的脂肪酸部分包括含量超过90%的ω-羟基化脂肪酸。由此获得的油性组合物的脂肪酸部分的重量百分比组成示于下文表1中。
[0145]
表1
[0146]
组分在提取物中的含量(%)p-香豆酸0.9十六烷酸2.04亚油酸0.46油酸0.28硬脂酸0.0516-羟基十六烷酸3.61,16-十六烷二酸0.6110,16-二羟基十六烷酸89.66羟基十六烷-1,16-二酸2.12二羟基辛烷酸0.28
[0147]
10,16-二羟基十六烷酸是该油性组合物的主要组分,占按重量计几乎90%的大多数分数。
[0148]
基于油性组合物和甘油的聚合物的制备
[0149]
将期望量的所得油性组合物引入到尺寸为4x4 cm、覆盖有特氟隆的不锈钢模具中。整体在烘箱(thermo scientific fb65500)中在60℃下预热5分钟。
[0150]
然后,将期望量的甘油引入到模具中,该量相对于甘油和油性组合物的混合物的总重量在按重量计5至25%之间变化,在这些实例中设定为1.8g。
[0151]
作为比较例,在仅用油性组合物而不添加甘油的情况下进行实验。
[0152]
实施的甘油的不同量示于下文表2中。
[0153]
表2-*脂肪酸的摩尔数是在考虑到油性组合物仅包含脂肪酸的情况下确定的,所有脂肪酸都具有与10,16-二羟基十六烷酸相同的摩尔质量
[0154][0155]
没有将其他化合物,特别是没有将催化剂或溶剂引入到反应介质中。
[0156]
将包含如此形成的反应介质的模具引入到60℃的烘箱中,并通过用刮刀手动搅拌5分钟将反应介质均化。还将干燥剂(氧化磷p2o5)引入到烘箱中。
[0157]
然后在持续90分钟的初始阶段在烘箱中施加150℃的温度和减压。在反应介质中油性组合物中包含的脂肪酸与甘油发生共聚,以及由此形成的聚合物开始交联。
[0158]
在不同的压力值0、200、400、600、800和1000mbar下进行不同的实验。
[0159]
在该初始阶段结束时,就在达到聚合物的凝胶点之前,烘箱中的压力恢复到大气压,并且通过每5分钟手动搅拌,持续20分钟来消除反应介质中形成的气泡。
[0160]
然后,在加热步骤的第二阶段期间,在大气压下,在150℃下加热22小时,以继续聚合物的交联。
[0161]
在该加热步骤完成之后,将模具从烘箱中移出并在冰浴中冷却。从中回收固体聚合物,对于用甘油的实例,其具有弹性变形特性。该聚合物具有这样的结构,其一个实例示于图1中。在脂肪酸的羧酸基团与羟基化脂肪酸和甘油的羟基之间形成了酯型键。
[0162]
初始试剂的酯化率分析
[0163]
对于每种形成的聚合物,确定酯化甘油的百分比和酯化脂肪酸的百分比。当化合物的至少一个官能团与聚合物内的酯键接合时,该化合物被认为是酯化的。
[0164]
为此,在室温下并且在搅拌下将5mg聚合物浸入1ml甲醇中16小时,以回收甘油和非酯化脂肪酸。对这种包含甘油和游离脂肪酸的甲醇级分的等分试样进行取样,并在甲硅烷基化后进样到gc-ms中。其次,使用弱碱(0.5m甲醇钠)在无水甲醇中解聚“洗涤过的”聚合物,即仅包含甘油和酯化脂肪酸。这种解聚在室温下在搅拌下进行16小时。为了完成解聚,将混合物在60℃下加热2小时。在甲硅烷基化后,对该级分的等分试样进行取样并进样到gc-ms中。由于内部标准,测定了不同的甘油和脂肪酸含量。
[0165]
所得结果(以作为初始反应介质中甘油的按重量计含量的函数的酯化产物百分比表示)示于图2(对于甘油)和图3(对于脂肪酸)中。观察到对于甘油和脂肪酸二者,聚合物中参与酯键的化合物的按重量计含量均高于85%。
[0166]
通过红外光谱对聚合物的分析
[0167]
通过傅里叶变换红外光谱(fourier-transform infrared spectroscopy,ftir)对已获得的聚合物以及初始油性组合物进行分析。
[0168]
为此,使用由软件控制的thermo scientific商业化的nicolet magna-ir550光谱仪。分辨率设置为2cm-1
,并且对每个光谱进行30次采集。
[0169]
对于初始油性组合物(ch)以及在400mbar减压下获得的例如pg25%聚合物(初始甘油含量为25%)的光谱示于图4中。图5示出了这些光谱区域的放大图。
[0170]
这些光谱清楚地显示了根据本发明形成的pg25%聚合物的聚酯性质。可以看出,初始油性组合物实际上呈现出以1705cm-1
为中心的强烈的单峰羰基带,这是酸的特征。对于pg25%弹性体,我们观察到该带在酯的波长特性1731cm-1
处的位移。酯羰基带在1715cm-1
处的肩部分裂,这反映了与(羟基化脂肪酸和/或甘油的)未酯化羟基形成氢键。初始油性组合物的光谱与pg25%弹性体的光谱之间在3500cm-1
处特征羟基带的降低进一步证实了羟基化脂肪酸的羟基的酯化。
[0171]
无论使用的甘油的初始含量如何,也无论施加的压力如何,对于所有制备的聚合物都获得了类似的光谱。
[0172]
这些结果特别证明,可以通过追踪相对于1705cm-1
处未酯化羰基带(羧酸盐)强度的1731cm-1
处酯羰基带强度来评估聚合水平并在时间-温度动力学过程中实时追踪聚合水平。
[0173]
图6示出了在所有受试压力值下,对于pg15%聚合物(初始甘油含量为15%)的特定实例,以羟基官能团的特征带为中心的这些光谱区域的放大图。我们观察到羟基特征带的强度随加热步骤期间施加的压力值而降低。这表明在装置中施加的压力越低,聚合物中的游离羟基官能团的数量就越少。1000mbar的压力给出了不能令人满意的未酯化羟基官能团水平。
[0174]
对于所有其他受试的初始甘油含量,获得了可比较的结果。
[0175]
酯化羟基的性质分析
[0176]
对于形成的每种聚合物,进一步确定了已经酯化的10,16-二羟基十六烷酸的羟基的相对比例。
[0177]
为此,如上文所解释的,应用philippe等在plant physiology,2016,170,807-820中的出版物中所描述的方法。将5mg聚合物与50mg 2-苄氧基-1-甲基吡啶三氟甲磺酸盐(sigma-aldrich)和6mg氧化镁在1ml三氟甲苯中于90℃下于塞紧的玻璃管中混合24小时。然后,聚合物用二氯甲烷清洗并干燥。然后,使用0.5m甲醇钠使标记的聚合物解聚。这种解聚在60℃下进行16小时。在硅烷化之后,对该级分的等分试样进行取样并进样到气相色谱与质谱联用(gc-ms)中。
[0178]
因此,对于所获得的每种聚合物,确定了对应于以下的片段的按重量计相对比例:下式(iiia)的在其两个羟基官能团上酯化的10,16-二羟基十六烷酸;下式(iiib)的仅在其仲羟基官能团上酯化的10,16-二羟基十六烷酸;下式(iiic)的仅在其伯羟基官能团上酯化的10,16-二羟基十六烷酸。
[0179][0180][0181]
对于在400mbar压力下形成的聚合物,所得结果示于图7中。观察到10,16-二羟基十六烷酸的羟基的酯化模式根据初始甘油重量含量而不同。与位于脂肪酸链中间的仲羟基相比,ω位置的伯羟基(即位于链末端)是最参与酯化/交联反应的那些。此外,对于由甘油获得的所有聚合物,其中两个羟基官能团被酯化的片段的比例低于70%。
[0182]
图8示出了对于pg15%聚合物在所有受试压力值下获得的结果。在0mbar的值下,其中两个羟基官能团被酯化的片段的比例高于80%,这是不令人满意的。对于400至800mbar的压力,获得了与聚合物最有利的机械特性相关的相对比例。
[0183]
对于每种形成的聚合物,还确定了已酯化的甘油羟基的相对比例。
[0184]
因此,对于每种获得的聚合物,确定了对应于以下的片段的按重量计相对比例:下式(iva)的在其三个羟基官能团上酯化的甘油;下式(ivb)的仅在1或3位的羟基官能团上酯化的甘油;下式(ivc)的仅在2位的羟基官能团上酯化的甘油。
[0185][0186]
对于在400mbar压力下获得的聚合物,所得结果示于图9中。观察到,对于由甘油获得的所有聚合物,其中所有羟基官能团都被酯化的来源于甘油的片段的比例在很大程度上占大多数。
[0187]
聚合物的玻璃化转变温度分析
[0188]
通过差示扫描量热法(differential scanning calorimetry,dsc)技术来测量所形成聚合物的玻璃化转变温度。
[0189]
为此,使用了dsc-q100热量计。将约10mg的样品放入密封的铝胶囊中。将这些胶囊冷却至-50℃,然后以3℃/分钟的升温斜率加热至80℃。
[0190]
对于在400mbar下获得的聚合物,所得结果(以作为反应介质中初始甘油重量含量的函数的由此测量的玻璃化转变温度表示)示于图10中。可以看出,根据本发明的聚合物的玻璃化转变温度为-12.5℃至-25℃,而对于在没有甘油的情况下形成的聚合物,其基本上等于-10℃。
[0191]
对于0至900mbar的其他受试压力值,获得了类似的结果。
[0192]
聚合物的机械弛豫温度分析
[0193]
使用还允许测量橡胶平台处的储能模量的rheometric scientificmk3e装置通过动态机械分析(dynamic mechanical analysis,dma)来确定所形成聚合物的机械弛豫温度。从-50℃至80℃,以3℃/分钟的升温速率、1hz的振荡频率、0.1%的应变和0.5n的预载,对每种聚合物的1mm厚、15mm高、4mm宽的样品进行了分析。
[0194]
对于在400mbar下获得的聚合物,所得结果(以作为反应介质中初始甘油重量含量的函数的由此测量的机械弛豫温度表示)示于图11中。对于0至900mbar的其他受试压力值,获得了类似的结果。
[0195]
聚合物的机械特性分析
[0196]
获得的聚合物在由4软件控制的mts拉伸台上进行拉伸测试。为此,制作具有图12所示形状和尺寸且厚度为1mm的试样。位移限定为10mm/分钟。测试在室温下进行。
[0197]
图13示出了作为反应介质中初始甘油重量含量的函数的在400mbar下所获得聚合物的测量的杨氏模量。这些结果表明,对于根据本发明的由甘油制备的所有聚合物,其杨氏模量低于对于不使用甘油制备的比较聚合物所测得的杨氏模量。
[0198]
图14示出了作为反应介质中初始甘油重量含量的函数的在400mbar下所获得聚合物的测量的最大拉伸强度。这些结果表明,对于根据本发明的由甘油制备的所有聚合物,其最大拉伸强度低于对于未实施甘油制备的比较聚合物所测得的最大拉伸强度。
[0199]
图15示出了作为反应介质中初始甘油重量含量的函数的在400mbar下所获得聚合物的测量的断裂伸长率百分比。这些结果表明,对于根据本发明的由甘油制备的所有聚合物,其断裂伸长率百分比远高于对于未实施甘油制备的比较聚合物所测得的断裂伸长率百分比。
[0200]
所有这些结果表明了根据本发明的聚合物的良好拉伸能力。
[0201]
对于0至900mbar的其他受试压力值,获得了类似的结果。
[0202]
溶解度测试
[0203]
对每种所获得聚合物的样品在以下不同溶剂中进行溶解度测试:水、甲醇、乙醇、异丙醇、氯仿、四氢呋喃、二氯甲烷、二甲亚砜。为此,将10mg每种样品浸入1ml溶剂中,然后将整体在室温下搅拌18小时。然后确定每个溶剂/样品对的不溶性物质相对于初始物质重量的按重量计百分比。
[0204]
作为一个实例,对于在加热步骤期间通过施加400mbar压力获得的pg5%聚合物所获得的结果显示在下表3中。
[0205]
表3
[0206]
溶剂不溶性物质的按重量计百分比(%)水94.5甲醇89.1乙醇91.1异丙醇94.1氯仿91.6四氢呋喃92.4二氯甲烷91二甲亚砜99.1
[0207]
对于根据本发明的其他聚合物,获得了类似的结果。
[0208]
结晶度分析
[0209]
根据上文所述的方案通过差示扫描量热法(dsc)对通过在加热步骤期间施加400mbar的压力获得的聚合物进行分析。pg0%聚合物的结果示于图16中,pg15%聚合物的结果示于图17中,pg25%聚合物的结果示于图18中。我们观察到,对于按重量计高于或等于15%的初始甘油重量含量,存在指示晶体结构的解链峰。
[0210]
这些结果通过配备有vantec 500检测器的bruker d8 x射线衍射仪对这些聚合物进行的x射线衍射(x-ray diffraction,xrd)分析得到证实。选择在40kv和40ma密封铜管中产生的x射线辐射cu kα1(=0.15406nm),并使用goebel反射镜平行化。样品与检测器之间的距离为10cm,并且布拉格角为3至70(
°
2θ)。
[0211]
对于pg0%聚合物获得的xrd光谱示于图19中,对于pg25%聚合物获得的xrd光谱示于图20中。对于pg25%聚合物,我们观察到出现指示结晶区的峰,如图中箭头所指示的。
[0212]
对于由反应介质中高于或等于15%的甘油重量含量获得的根据本发明的其他聚合物,获得了类似结果。
[0213]
图21示出了对于通过在加热步骤期间施加400mbar或1000mbar的压力,由反应介质中重量含量为15%的甘油制备的聚合物获得的xrd光谱。观察到对于根据本发明的400mbar的压力获得了半结晶结构。
[0214]
图22示出了对于以下获得的drx光谱:初始番茄角质提取物(在a/中);以及由该提取物和重量含量为20%的甘油在加热步骤期间施加了400mbar的压力下获得的聚合物(在b/中)。清楚地观察到根据本发明制备的聚合物的结晶组织不同于初始提取物的结晶组织。在油性组合物的光谱上观察到的指示β结晶的峰已经消失,而在聚合物光谱上,在4.2在六角相中出现了指示结晶的峰。
[0215]
橡胶平台上的储能模量和表观交联密度的分析
[0216]
如前所述,储能模量是借助于rheometric scientific mk3e装置通过动态机械分析(dma)测量的。
[0217]
在50℃的温度下,该材料的行为类似于橡胶,其储能模量在兆帕斯卡范围内。然后flory橡胶弹性理论的应用使得能够根据下式估计弹性体网络的ν表观交联密度:
[0218]
v=e
′
/3rt
[0219]
其中r为气体常数(8.32j.mol-1
.k-1
),t为开尔文温度,并且e
′
为在50℃(323k)下测量的橡胶平台上的储能模量。
[0220]
对于400mbar的压力获得的结果作为初始甘油重量含量的函数显示在图23中。观察到,在50℃下,在非晶态橡胶平台上测得的储能模量e
′
随反应介质中初始甘油含量的增加而降低,然后在甘油含量按重量计高于或等于15%时变得恒定。表观交联密度也是如此,根据flory理论表观交联密度与弹性模量成正比。
[0221]
这些结果证明根据本发明的方法允许生产具有受控分子结构和具有非晶态或半结晶性质的聚合物,这取决于初始反应介质中甘油的重量含量。
[0222]
对于由按重量计15%的初始甘油含量获得的聚合物,图24中示出了对于在该方法的加热步骤期间施加的不同压力值测得的储能模量与在大气压下实施该方法时测得的储能模量的比值(e
′
/eatm比值)。该比值在400至800mbar的压力范围内基本恒定,这表明根据本发明在该范围内获得的聚合物具有稳定结构。
[0223]
解链温度的测定
[0224]
解链温度通过差示焓分析(dea或差示扫描量热法(dsc))测量,包括对聚合物样品进行从-50℃温度至80℃温度的温度扫描,升温斜率为3℃/分钟。该分析是对置于密封铝胶囊中的10mg量的聚合物进行的。
[0225]
以下表4和表5中报告了作为一个实例对于根据本发明在加热步骤期间施加400mbar压力形成的聚合物所获得的所有结果。
[0226]
表4-根据本发明在加热期间施加了400mbar压力下获得的聚合物的化学和dsc表征
[0227][0228]
表5-根据本发明在加热期间施加了400mbar压力下获得的聚合物通过dma和通过机械测试的表征
[0229][0230]
所有这些结果都特别令人满意,尤其是pg 15%、pg20%和pg25%聚合物。
[0231]
实施例2-苹果角质
[0232]
从来源于苹果转化的工业果渣中,在与实施例1中对于番茄所描述的相同的条件下提取了苹果角质的羟基化脂肪酸。
[0233]
在用脱矿质水清洗获得的沉淀物并冷冻干燥后,获得油性组合物,产率为20至25%,该油性组合物基本上包含羟基化脂肪酸,并且更特别地,以相对于油性组合物总重量的重量百分比表示:二羟基十六烷酸44%、18-羟基-(9,10)环氧-十八烷酸24%、18-羟基-十八碳烯酸10%、三羟基十八烷酸7%、16-羟基十六烷酸5%、棕榈酸6%、十六烷二酸2%、22-羟基-二十二烷酸1%、香豆酸1%。
[0234]
基于该油性组合物和甘油,在实施例1中所描述的条件下,在150℃的加热温度、400mbar的减压和10%(w/w)的甘油含量下制备聚合物。
[0235]
聚酯的形成通过如实施例1所述进行的傅里叶变换红外光谱分析得到证实:如图
25所示,我们观察到,在红外光谱上,相对于初始油性组合物(“chp”),对于所获得的聚合物(“ppg10%”),出现酯基团的带特征和羰基带特征的位移。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。