1.本发明属于医药技术领域。特别涉及通式(i)所示的芳基巴比妥类化合物,其制备方法,以该化合物为活性成分的药物组合物,它们在制备结核分枝杆菌酪氨酸磷酸酶抑制剂中的应用,以及它们在治疗和/或预防由结核分枝杆菌引起的感染性疾病中的应用。
背景技术:
2.结核病(tuberculosis,tb)是由结核分枝杆菌引起的一种慢性致死性疾病,是危害人类健康和导致人类死亡的重大传染性疾病。世界卫生组织(who)报告显示:2018年,全世界预计有新发结核病患者1000万,死亡患者120万,是目前致死率排名前十的疾病之一,其致死率高于艾滋病。在2018年的结核病患者中,约50万为一线药利福平耐药(rr-tb)。
3.中国的结核病形势非常严峻,2018年肺结核发病数近83万仅次于病毒性肝炎位居甲乙类传染病的第二位。世界范围的mdr-tb发病几乎一半发生在印度(24%)、中国(13%)和俄罗斯(10%)。因此,研发具有新结构和新机制,能够对抗mdr-tb和xdr-tb的抗结核药物具有十分重要的现实意义和社会意义。
4.结核分枝杆菌侵染人体后,被肺部巨噬细胞中的吞噬体吞噬,与此同时,初始免疫反应启动,免疫反应细胞将受感染的巨噬细胞隔离,形成肉芽肿,肉芽肿含有大量血管以及免疫细胞,这提高了药物到达感染部位和宿主免疫系统抵抗病原体的能力。然而结核分枝杆菌感染巨噬细胞后会分泌蛋白酪氨酸磷酸酶b(mycobacterium tuberculosis protein tyrosine phosphatase b,mptpb),mptpb使宿主信号转导过程中的蛋白去磷酸化,调控宿主与病原体相互作用,阻止了巨噬细胞免疫系统的启动,结核分枝杆菌依靠这些方式逃避免疫系统的杀灭。
5.抑制mptpb,可恢复宿主正常功能和免疫能力进而杀灭结核分枝杆菌。mptpb抑制剂作用机制独特,不需要进入结核分枝杆菌内部即可发挥抗结核作用,具有克服耐药、缩短治疗周期等优点,这为有效治疗结核病,特别是提高耐药结核病的治愈率、治疗免疫功能低下或障碍的结核病患者(如hiv感染者、器官移植患者),提供了新的治疗策略。
6.文献(bioorganic chemistry,2019,85,229-239)报道了如式(v)所示化合物,其在50μm时,对mptpb抑制率为70%,其mptpb抑制活性ic
50
=22.4μm,表明该化合物对mptpb抑制活性弱。
7.
技术实现要素:
8.本发明要解决的技术问题是提供一种结构新颖、mptpb抑制活性强的芳基巴比妥类化合物。
9.为此,本发明第一方面提供通式(i)所示的化合物或其药学上可接受的盐,
[0010][0011]
其中,
[0012]
为碳碳单键或碳碳双键;
[0013]
x选自o或s;
[0014]
r1、r2独立地选自氢、c
1-c4烷基、c
1-c2烷基oc
1-c2烷基或卤代c
1-c4烷基;
[0015]
r3选自取代或未取代的苯基、取代或未取代的5-6元杂芳基;
[0016]
r4选自氢、取代或未取代的c
1-c4烷基、取代或未取代的c
3-c6环烷基、取代或未取代的苯基、取代或未取代的5-6元杂芳基;
[0017]
所述r3、r4中取代或未取代的5-6元杂芳基至少含有一个选自n、o、s中的杂原子;
[0018]
所述r3、r4中取代或未取代的取代基独立地选自以下基团:f、cl、br、羟基、羧基、-cooch3、硝基、氰基、三氟甲基、三氟甲氧基、c
1-c4烷基、c
3-c6环烷基、c
1-c3烷氧基或c
1-c3烷胺基;
[0019]
基团与苯环的1、2、3或4位相连。
[0020]
在一些方面,式(i)所示化合物:
[0021]
其中,
[0022]
为碳碳单键或碳碳双键;
[0023]
x选自o或s;
[0024]
r1、r2独立地选自氢、c
1-c4烷基、c
1-c2烷基oc
1-c2烷基或卤代c
1-c4烷基;
[0025]
r3为
[0026]
其中r5、r6、r7和r8独立地选自以下的一个或多个取代基:f、cl、br、羟基、羧基、-cooch3、硝基、氰基、三氟甲基、三氟甲氧基、c
1-c4烷基、c
3-c6环烷基、c
1-c3烷氧基或c
1-c3烷胺基;
[0027]
r4为氢、
[0028]
其中r9、r
10
、r
11
、r
12
和r
13
独立地选自以下的一个或多个取代基:f、cl、br、硝基、羟基、氰基、三氟甲基、三氟甲氧基、c
1-c4烷基、c
3-c6环烷基、c
1-c3烷氧基或c
1-c3烷胺基。
[0029]
基团与苯环的1、2、3或4位相连。
[0030]
在一些方面,式(i)所示化合物:
[0031]
其中,
[0032]
为碳碳单键或碳碳双键;
[0033]
x选自o或s;
[0034]
r1、r2独立地选自氢、c
1-c4烷基、c
1-c2烷基oc
1-c2烷基或卤代c
1-c4烷基;
[0035]
r3为为
[0036]
r4为氢、
[0037]
基团与苯环的1、2或3位相连。
[0038]
在一些方面,式i化合物选自式(ii)化合物:
[0039][0040]
其中,
[0041]
为碳碳单键或碳碳双键;
[0042]
x选自o或s;
[0043]
r1、r2独立地选自氢、c
1-c4烷基、c
1-c2烷基oc
1-c2烷基或卤代c
1-c4烷基;
[0044]
r3为
[0045]
r4为氢、
[0046]
在一些方面,式i化合物选自式(iii):
[0047][0048]
其中,
[0049]
为碳碳单键或碳碳双键;
[0050]
x选自o或s;
[0051]
r1、r2独立地选自氢、c
1-c4烷基、c
1-c2烷基oc
1-c2烷基或卤代c
1-c4烷基;
[0052]
r3为为
[0053]
r4为氢、
[0054]
在一些方面,式i化合物选自式(iv)化合物:
[0055][0056]
其中,
[0057]
为碳碳单键或碳碳双键;
[0058]
x选自o或s;
[0059]
r1、r2独立地选自氢、c
1-c4烷基、c
1-c2烷基oc
1-c2烷基或卤代c
1-c4烷基;
[0060]
r3为为
[0061]
r4为氢、
[0062]
根据本发明第一方面任一项化合物,其为选自下列的化合物:
[0063]
[0064]
[0065]
[0066][0067][0068]
本发明第二方面提供了制备本发明第一方面任一项所述化合物的方法,其包括以
下步骤:
[0069]
(1)
[0070][0071]
化合物a通过williamson合成法得到化合物b,化合物b通过miyaura硼酸酯化反应得到化合物c,化合物c通过suzuki偶联反应得到化合物d,化合物d通过knoevenagel缩合反应得到式(i)所示碳碳双键化合物,其碳碳双键经还原后得到式(i)所示碳碳单键化合物;
[0072]
或(2)
[0073][0074]
化合物a通过williamson合成法或mitsunobu反应得到化合物b,化合物b通过suzuki偶联反应得到化合物d,化合物d通过knoevenagel缩合反应得到式(i)所示碳碳双键化合物,其碳碳双键经还原后得到式(i)所示碳碳单键化合物。
[0075]
r1、r2、r3、r4和x定义同上述限定。
[0076]
本发明第三方面提供了一种药用组合物,其包括治疗和/或预防有效量的第一方面任一项所述的化合物或其药学上可接受的盐以及任选的一种或多种药学上可接受的载体、赋形剂、稀释剂、辅料和媒介物。
[0077]
本发明第四方面提供了本发明第一方面任一项所述化合物及其药学可接受的盐,或者本发明第三方面任一项所述药物组合物在制备结核分枝杆菌酪氨酸磷酸酶抑制剂中的应用。
[0078]
本发明第五方面提供了本发明第一方面任一项所述化合物及其药学可接受的盐,
或者本发明第三方面任一项所述药物组合物在制备治疗和/或预防由结核分枝杆菌引起的感染性疾病的药物中的应用。
[0079]
发明详述
[0080]
下面对本发明的各个方面和特点作进一步的描述。
[0081]
本发明所引述的所有文献,它们的全部内容通过引用并入本文,并且如果这些文献所表达的含义与本发明不一致时,以本发明的表述为准。此外,本发明使用的各种术语和短语具有本领域技术人员公知的一般含义,即便如此,本发明仍然希望在此对这些术语和短语作更详尽的说明和解释,提及的术语和短语如有与公知含义不一致的,以本发明所表述的含义为准。下面是本发明所用多种术语的定义,这些定义适用于本技术整个说明书中所用的术语,除非在具体情况中另作说明。
[0082]
术语“取代的”是指所给结构中特定原子上的任意一个或多个氢原子被具体取代基所取代,只要特定原子的价态是正常的并且取代后所得化合物是稳定的。除非其他方面表明,一个任选的取代基团可以在基团各个可取代的位置进行取代。当给出的结构式中不只一个位置能被选自具体基团的一个或多个取代基所取代,那么取代基可以相同或不同地在各个位置取代。
[0083]
各种含烃部分的碳原子含量通过指明了该部分中最小和最大碳原子数的前缀表示。c
i-cj表示具有整数“i”(包含i)至整数“j”(包含j)个碳原子的部分。因此,例如c
1-c4烷基指具有1至4个(包含1和4)碳原子的烷基,特别指甲基、乙基、c3烷基和c4烷基。
[0084]
如本文所述的,术语“烷基”是指具有指定数目碳原子数的烷基,其为直链或支链的烷基,并且其可包括其子基团,例如提及“c
1-c4烷基”时,其还可以包括c
1-c3烷基、c
1-c2烷基、c
2-c4烷基、c
3-c4烷基等表示的子范围的基团,以及具体基团例如甲基、乙基、正丙基、异丙基。术语“烷氧基”和“烷胺基”属于惯用表达,是指分别通过一个氧原子或胺基连接到分子的其余部分的烷基基团,其中的烷基如本发明所述。烷氧基基团包括,但并不限于,甲氧基、乙氧基、异丙氧基、正丙氧基、等等。烷胺基基团包括,但并不限于,甲胺基、乙胺基、异丙胺基、正丙胺基、等。
[0085]
术语“卤代烷基”表示烷基基团被一个或多个卤素原子所取代,包含,但并不限于,三氟甲基、二氟甲基等。
[0086]
如本文所述的,术语“卤”、“卤代”等表示氟(f)、氯(cl)、溴(br)或碘(i)。
[0087]
如本文所述的,术语“环烷基”是指具有指定数目环碳原子数的环状烷基,并且其可包括其子基团,例如提及“3-6元环烷基”时,其还可以包括3-5元环烷基、4-6元环烷基等表示的子范围的基团,以及具体基团例如环丙基、环丁基、环戊基、环己基。
[0088]
如本文所述的,术语“杂芳基”在本文中指具有1至3个杂原子作为环原子,其余的环原子为碳的芳香基团,其中杂原子包括氧、硫和氮。例如“5-6元杂芳基”包括5元杂芳基和6元杂芳基。其中5元杂芳基包括,但并不限于咪唑基、呋喃基、噻吩基、三氮唑基、四氮唑基、吡唑基(如2-吡唑基)、噻唑基、噁唑基、异恶唑基。6元杂芳基包括吡啶基、哒嗪基、嘧啶基、吡嗪基、1,3,5-三嗪基。在实施方案中,所述的杂芳基为噻吩基、呋喃基、吡啶基。
[0089]
如本文所述的,术语“环”表示被取代或未被取代的环烷基、被取代或未被取代的杂环基、被取代或未被取代的芳基或被取代或未被取代的杂芳基。所谓的环包括稠环。环上原子的数目通常被定义为环的元数,例如“3-6元环”是指环绕排列3-6个原子。
[0090]
如本文所述的,术语“有效量”是指可在受试者中实现所期望的治疗本发明所述疾病或病症的药物用量。
[0091]
如本文所述的,术语“药学可接受的”例如在描述“药学可接受的盐”时,表示该盐不但是受试者生理学上可接受,而且还可指在药学上有使用价值的合成物质,例如在为进行手性拆分时所形成的作为中间体的盐,虽然这种中间体的盐并不能直接给予受试者,但该盐可在为获得本发明终产物中起作用。
[0092]
如本文所述的,术语“药物组合物”,其还可以是指“组合物”,其可用于在受试者特别是哺乳动物中实现治疗本发明所述疾病或病症。
[0093]
疾病的“治疗”包括:
[0094]
(1)预防该疾病,即,使暴露至或易感染该疾病但未经历或显示该疾病症状的哺乳动物不发生该疾病的临床症状,
[0095]
(2)抑制该疾病,即,阻止或减少该疾病或其临床症状的进展,
[0096]
(3)减轻该疾病,即,引起该疾病或其临床症状的复原。
[0097]
如本文所述的,术语“疾病和/或病症”是指所述受试者的一种身体状态,该身体状态与本发明所述疾病和/或病症有关。例如,本发明所述疾病和/或病症指结核分枝杆菌感染性疾病。
[0098]
本发明再一方面还涉及以本发明中的化合物作为活性成份的药物组合物。该药物组合物可根据本领域公知的方法制备。可通过将本发明化合物与一种或多种药学上可接受的固体或液体赋形剂和/或辅剂结合,制成适于人或动物使用的任何剂型。
[0099]
本发明中的化合物或含有它的药物组合物可以单位剂量形式给药,给药途径可为肠道或非肠道,如口服、静脉注射、肌肉注射、皮下注射、鼻腔、口腔粘膜、眼、肺和呼吸道、皮肤、阴道、直肠等。
[0100]
给药剂型可以是液体剂型、固体剂型或半固体剂型。液体剂型可以是溶液剂(包括真溶液和胶体溶液)、乳剂(包括o/w型、w/o型和复乳)、混悬剂、注射剂(包括水针剂、粉针剂和输液)、滴眼剂、滴鼻剂、洗剂和搽剂等;固体剂型可以是片剂(包括普通片、肠溶片、含片、分散片、咀嚼片、泡腾片、口腔崩解片)、胶囊剂(包括硬胶囊、软胶囊、肠溶胶囊)、颗粒剂、散剂、微丸、滴丸、栓剂、膜剂、贴片、气(粉)雾剂、喷雾剂等;半固体剂型可以是软膏剂、凝胶剂、糊剂等。
[0101]
本发明化合物可以制成普通制剂、也可以制成缓释制剂、控释制剂、靶向制剂及各种微粒给药系统。
[0102]
为了将本发明化合物制成片剂,可以广泛使用本领域公知的各种赋形剂,包括稀释剂、黏合剂、润湿剂、崩解剂、润滑剂、助溶剂。稀释剂可以是淀粉、糊精、蔗糖、葡萄糖、乳糖、甘露醇、山梨醇、木糖醇、微晶纤维素、硫酸钙、磷酸氢钙、碳酸钙等;湿润剂可以是水、乙醇、异丙醇等;粘合剂可以是淀粉浆、糊精、糖浆、蜂蜜、葡萄糖溶液、微晶纤维素、阿拉伯胶浆、明胶浆、羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、乙基纤维素、丙烯酸树脂、卡波姆、聚乙烯吡咯烷酮、聚乙二醇等;崩解剂可以是干淀粉、微晶纤维素、低取代羟丙基纤维素、交联聚乙烯吡咯烷酮、交联羧甲基纤维素钠、羧甲基淀粉钠、碳酸氢钠与枸橼酸、聚氧乙烯山梨糖醇脂肪酸酯、十二烷基磺酸钠等;润滑剂和助溶剂可以是滑石粉、二氧化硅、硬脂酸盐、酒石酸、液体石蜡、聚乙二醇等。
[0103]
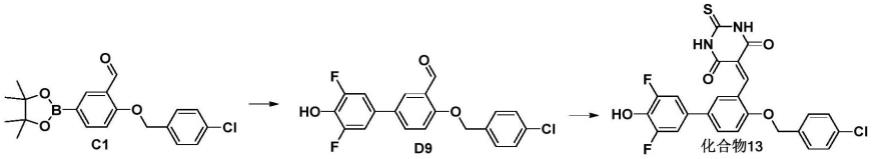
还可以将片剂进一步制成包衣片,例如糖包衣片、薄膜包衣片、肠溶包衣片,或双层片和多层片。
[0104]
为了将给药单元制成胶囊剂,可以将有效成分本发明化合物与稀释剂、助溶剂混合,将混合物直接置于硬胶囊或软胶囊中。也可将有效成分本发明化合物先与稀释剂、黏合剂、崩解剂制成颗粒或微丸,再置于硬胶囊或软胶囊中。用于制备本发明化合物片剂的各稀释剂、黏合剂、润湿剂、崩解剂、助溶剂品种也可用于制备本发明化合物的胶囊剂。
[0105]
为将本发明化合物制成注射剂,可以用水、乙醇、异丙醇、丙二醇或它们的混合物作溶剂并加入适量本领域常用的增溶剂、助溶剂、ph调节剂、渗透压调节剂。增溶剂或助溶剂可以是泊洛沙姆、卵磷脂、羟丙基-β-环糊精等;ph调节剂可以是磷酸盐、醋酸盐、盐酸、氢氧化钠等;渗透压调节剂可以是氯化钠、甘露醇、葡萄糖、磷酸盐、醋酸盐等。如制备冻干粉针剂,还可加入甘露醇、葡萄糖等作为支撑剂。
[0106]
此外,如需要,也可以向药物制剂中添加着色剂、防腐剂、香料、矫味剂或其它添加剂。
[0107]
为达到用药目的,增强治疗效果,本发明的药物或药物组合物可用任何公知的给药方法给药。
[0108]
本发明的化合物或组合物可单独服用,或与其他治疗药物或对症药物合并使用。当本发明的化合物与其它治疗药物存在协同作用时,应根据实际情况调整它的剂量。
[0109]
有益技术效果
[0110]
本发明人发现,本发明的化合物具有非常强的mptpb抑制活性。文献(bioorganic chemistry,2019,85,229-239)报道的化合物(v),其在50μm浓度时,对mptpb抑制率为70%,其mptpb抑制活性ic
50
=22.4μm,活性弱。当化合物(v)中苯环上的br以芳基取代时,化合物对mptpb抑制活性显著增加,在50μm浓度时,对mptpb抑制率大于70%。本发明中化合物2表现出非常强的mptpb抑制活性,在50μm浓度时,mptpb抑制率为95.91%,其mptpb抑制活性ic
50
=1.18μm。本发明化合物2与对比文献中化合物v相比,mptpb抑制活性明显增强,具有显著的进步。本发明提供了一类结构新颖、活性强、毒性低的芳基巴比妥类化合物,该类化合物可用于由结核分枝杆菌引起的感染性疾病的预防和治疗。
[0111]
具体实施方式
[0112]
通过下面的实施例可以对本发明进行详细描述,但并不意味着对本发明任何不利限制。本文已经详细地描述了本发明,其中也公开了其具体实施例,对本领域的技术人员而
言,在不脱离本发明精神和范围的情况下针对本发明具体实施方式进行各种变化和改进是显而易见的。
[0113]
对于以下全部实施例,可使用本领域技术人员已知的标准操作和纯化方法。除非另有说明,所有温度以℃(摄氏度)表示。化合物的结构是通过核磁共振氢谱(1h nmr)来确定的。核磁共振氢谱位移(δ)以百万分之一(ppm)的单位给出。耦合常数(j)以赫兹(hz)为单位。核磁共振谱用mercury-400或brucker-500型核磁共振仪测定,氘代丙酮或氘代二甲基亚砜作溶剂,四甲基硅烷(tms)为内标。
[0114]
实施例1
[0115][0116]
5-((3',5'-二氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1'-联苯基]-3-基)-亚甲基)嘧啶-2,4,6(1h,3h,5h)-三酮(化合物1)的制备
[0117]
合成路线:
[0118][0119]
实验步骤:
[0120]
第一步5-溴-2-((4-氯苄基)氧基)苯甲醛b1的制备
[0121]
于100ml反应瓶中加入化合物a1(6.03g,30mmol)、4-氯苄氯(5.8g,36mmol)、碳酸钾(8.3g,60mmol),并用氩气保护,注入n,n-二甲基甲酰胺(50ml),120-125℃加热4小时。冷却,加入水(100ml),可析出固体,过滤,水洗,干燥得类白色固体9.62g,收率98.5%。
[0122]1h nmr(400mhz,dmso-d6)δ10.33(s,1h),7.83(dd,j=9.2,2.6hz,1h),7.77(d,j=2.6hz,1h),7.57
–
7.53(m,2h),7.50
–
7.46(m,2h),7.31(d,j=9.2hz,1h),5.31(s,2h).
[0123]
第二步3',5'-二氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1'-联苯基]-3-甲醛d1的制备
[0124]
于100ml反应瓶中加入化合物b1(326mg,1mmol)、3,5-二氯-4-羟基苯硼酸频哪醇酯(3.47mg,1.2mmol)、四(三苯基膦)钯(36mg,0.03mmol)、碳酸钠(318mg,3mmol),并用氩气保护,注入1,4-二氧六环(7.5ml)和水(1.5ml),105℃加热6小时。冷却,加入1mol/l盐酸
(15ml),用乙酸乙酯(15ml)萃取3次,合并有机层。有机层用饱和食盐水洗1次,用无水硫酸钠干燥,过滤,浓缩,硅胶(200-300目)柱色谱分离,石油醚-二氯甲烷(v:v=100:40~60)混合液为洗脱剂。得化合物d1,类白色固体301mg,收率73.8%。
[0125]1h nmr(400mhz,dmso-d6)δ10.43(s,1h),10.27(s,1h),7.98(dd,j=8.8,2.6hz,1h),7.93(d,j=2.6hz,1h),7.68(s,2h),7.60
–
7.55(m,2h),7.52
–
7.45(m,2h),7.37(d,j=8.8hz,1h),5.35(s,2h).
[0126]
第三步5-((3',5'-二氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1'-联苯基]-3-基)-亚甲基)嘧啶-2,4,6(1h,3h,5h)-三酮(化合物1)的制备
[0127]
于25ml反应瓶中加入化合物d1(61mg,0.15mmol)、嘧啶-2,4,6(1h,3h,5h)-三酮二水合物(25mg,0.15mmol),注入无水乙醇(6ml),加入浓硫酸(1滴),95℃加热5小时。趁热抽滤,滤饼依次用热水(3ml
×
3)、无水乙醇(3ml
×
3)洗,干燥。得化合物1,黄色固体55mg,收率70.5%。
[0128]1h nmr(400mhz,dmso-d6)δ11.37(s,1h),11.24(s,1h),10.24(s,1h),8.53(s,1h),8.33(d,j=2.2hz,1h),7.81(dd,j=8.8,2.2hz,1h),7.62(s,2h),7.51
–
7.46(m,4h),7.23(d,j=8.8hz,1h),5.29(s,2h).
[0129]
实施例2
[0130][0131]
5-((3',5'-二氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物2)的制备
[0132]
以化合物d1(612mg,1.5mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(216mg,1.5mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物2,橘黄色固体626mg,收率78.2%。
[0133]1h nmr(400mhz,dmso-d6)δ12.43(s,1h),12.33(s,1h),10.25(s,1h),8.55(s,1h),8.39(d,j=2.4hz,1h),7.84(dd,j=8.8,2.4hz,1h),7.63(s,2h),7.52
–
7.46(m,4h),7.24(d,j=8.8hz,1h),5.30(s,2h).
[0134]
实施例3
[0135][0136]
5-((3',5'-二氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-1,3-二甲基嘧啶-2,4,6(1h,3h,5h)-三酮(化合物3)的制备
[0137]
于10ml反应瓶中加入化合物d1(61mg,0.15mmol)、1,3-二甲基嘧啶-2,4,6(1h,3h,
5h)-三酮(23mg,0.15mmol),注入无水甲醇(2ml),加入浓硫酸(1滴),65℃加热3小时。趁热抽滤,滤饼依次用热水(3ml
×
3)、无水甲醇(3ml
×
3)洗,干燥。得化合物3,黄色固体37mg,收率45.1%。
[0138]1h nmr(400mhz,dmso-d6)δ10.25(s,1h),8.56(s,1h),8.19(d,j=2.4hz,1h),7.80(dd,j=8.8,2.4hz,1h),7.61(s,2h),7.50-7.45(m,4h),7.24(d,j=8.8hz,1h),5.27(s,2h),3.23(s,3h),3.13(s,3h).
[0139]
实施例4
[0140][0141]
5-((3',5'-二氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物4)的制备
[0142]
以化合物d1(122mg,0.3mmol)和1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(52mg,0.3mmol)为原料,采用实施例3中相似操作步骤,得到化合物4,橘黄色固体136mg,收率81.0%。
[0143]1h nmr(400mhz,dmso-d6)δ10.25(s,1h),8.60(s,1h),8.26(d,j=2.4hz,1h),7.84(dd,j=8.8,2.4hz,1h),7.64(s,2h),7.50-7.46(m,4h),7.25(d,j=8.8hz,1h),5.29(s,2h),3.65(s,3h),3.52(s,3h).
[0144]
实施例5
[0145][0146]
5-((3',5'-二氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-1-甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物5)的制备
[0147]
于10ml封管中加入化合物d1(82mg,0.2mmol)、1-甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(32mg,0.2mmol),注入甲苯(2ml),加入冰醋酸(2滴),微波135℃加热2小时。冷却,加入正己烷(5ml)稀释,减压抽滤,滤饼用正己烷(5ml
×
5)洗,干燥,再用正己烷-乙酸乙酯(6ml:1d)打浆,减压抽滤,干燥。得化合物5(e/z=3:4or 4:3),橘色固体86mg,收率78.2%。
[0148]1h nmr(400mhz,dmso-d6)δ12.57/12.49(s,1h),10.26/10.25(s,1h),8.61/8.53(s,1h),8.41/8.27(d,j=2.4hz,1h),7.86/7.82(dd,j=8.8,2.4hz,1h),7.63/7.62(s,2h),7.52
–
7.45(m,4h),7.26/7.23(d,j=8.8hz,1h),5.30/5.29(s,2h),3.57/3.46(s,3h).
[0149]
实施例6
[0150][0151]
5-((4-((4-氯苄基)氧基)-4'-甲氧基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物6)的制备
[0152]
合成路线:
[0153][0154]
第一步4-((4-氯苄基)氧基)-4'-甲氧基-[1,1
’‑
联苯基]-3-甲醛d2的制备
[0155]
于100ml反应瓶中加入化合物b1(880mg,2.7mmol)、4-甲氧基苯硼酸(451mg,2.97mmol)、醋酸钯(18mg,0.081mmol)、三苯基膦(106mg,0.405mmol)、碳酸钠(1.15g,10.8mmol),并用氩气保护,注入甲苯(15ml)、无水乙醇(6ml)和水(5.5ml),105℃加热5小时。冷却,加入水(30ml),用乙酸乙酯(30ml)萃取3次,合并有机层。有机层用饱和食盐水洗1次,用无水硫酸钠干燥,过滤,浓缩,硅胶(200-300目)柱色谱分离,石油醚-乙酸乙酯(v:v=100:15~50)混合液为洗脱剂。得化合物d2,黄色固体913mg,收率95.8%。
[0156]1h nmr(400mhz,dmso-d6)δ10.46(s,1h),7.94
–
7.89(m,2h),7.61
–
7.56(m,4h),7.51
–
7.46(m,2h),7.39
–
7.35(m,1h),7.04
–
6.99(m,2h),5.34(s,2h),3.79(s,3h).
[0157]
第二步5-((4-((4-氯苄基)氧基)-4'-甲氧基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物6)的制备
[0158]
以化合物d2(529mg,1.5mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(216mg,1.5mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物6,橘色固体604mg,收率84.1%。
[0159]1h nmr(400mhz,dmso-d6)δ12.42(s,1h),12.29(s,1h),8.61(s,1h),8.42(d,j=2.4hz,1h),7.78(dd,j=8.8,2.4hz,1h),7.58
–
7.54(m,2h),7.52
–
7.46(m,4h),7.24(d,j=8.8hz,1h),7.04
–
7.00(m,2h),5.30(s,2h),3.79(s,3h).
[0160]
实施例7
[0161][0162]
5-((4-((4-氯苄基)氧基)-4'-三氟甲基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物7)的制备
[0163]
合成路线:
[0164][0165]
第一步4-((4-氯苄基)氧基)-4'-三氟甲基-[1,1
’‑
联苯基]-3-甲醛d3的制备
[0166]
将实施例6中第一步中的4-甲氧基苯硼酸替换为4-三氟甲基苯硼酸,进行反应,可得化合物d3,白色固体659mg,收率71.5%。
[0167]1h nmr(400mhz,dmso-d6)δ10.47(s,1h),8.09
–
8.04(m,2h),7.91(d,j=8.0hz,2h),7.81(d,j=8.0hz,2h),7.59(d,j=8.4hz,2h),7.49(d,j=8.4hz,2h),7.45(d,j=8.8hz,1h),5.38(s,2h).
[0168]
第二步5-((4-((4-氯苄基)氧基)-4'-三氟甲基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物7)的制备
[0169]
以化合物d3(586mg,1.5mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(216mg,1.5mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物7,黄色固体334mg,收率43.1%。
[0170]1h nmr(500mhz,dmso-d6)δ12.45(s,1h),12.32(s,1h),8.59(s,1h),8.51(d,j=2.0hz,1h),7.92(dd,j=9.0hz,j=2.0hz,1h),7.86(d,j=8.0hz,2h),7.82(d,j=8.0hz,2h),7.52(d,j=8.0hz,2h),7.48(d,j=8.0hz,2h),7.32(d,j=9.0hz,1h),5.33(s,2h).
[0171]
实施例8
[0172][0173]
5-((4'-氯-4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物8)的制备
[0174]
合成路线:
[0175][0176]
第一步4-((4-氯苄基)氧基)-4'-氯-[1,1
’‑
联苯基]-3-甲醛d4的制备
[0177]
将实施例6中第一步中的4-甲氧基苯硼酸替换为4-氯苯硼酸,进行反应,可得中间体d4,类白色固体778mg,收率80.6%。
[0178]1h nmr(400mhz,dmso-d6)δ10.46(s,1h),7.80
–
7.96(m,2h),7.72
–
7.68(m,2h),7.61
–
7.55(m,2h),7.53
–
7.46(m,4h),7.41(d,j=8.6hz,1h),5.36(s,2h).
[0179]
第二步5-((4'-氯-4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物8)的制备
[0180]
以化合物d4(536mg,1.5mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(216mg,1.5mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物8,黄色固体511mg,收率70.5%。
[0181]1h nmr(400mhz,dmso-d6)δ12.44(s,1h),12.31(s,1h),8.59(s,1h),8.45(d,j=2.4hz,1h),7.84(dd,j=8.8,2.4hz,1h),7.67
–
7.64(m,2h),7.54
–
7.45(m,6h),7.28(d,j=8.8hz,1h),5.31(s,2h).
[0182]
实施例9
[0183][0184]
5-((4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物9)的制备
[0185]
合成路线:
[0186][0187]
第一步4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-甲醛d5的制备
[0188]
将实施例1中第二步中的3,5-二氯-4-羟基苯硼酸频哪醇酯替换为苯硼酸,进行反应,可得中间体d5,类白色固体777mg,收率80.3%。
[0189]1h nmr(400mhz,dmso-d6)δ10.47(s,1h),7.99
–
7.96(m,2h),7.68
–
7.64(m,2h),7.58
–
7.57(m,2h),7.50
–
7.45(m,4h),7.42
–
7.39(m,1h),7.39
–
7.34(m,1h),5.35(s,2h).
[0190]
第二步5-((4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物9)的制备
[0191]
以化合物d5(161mg,0.5mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(72mg,0.5mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物9,黄色固体174mg,收率77.7%。
[0192]1h nmr(400mhz,dmso-d6)δ12.44(s,1h),12.30(s,1h),8.61(s,1h),8.47(d,j=2.0hz,1h),7.84(dd,j=8.8,2.0hz,1h),7.64(d,j=7.2hz,2h),7.53
–
7.43(m,6h),7.35(t,j=7.2hz,1h),7.28(d,j=8.8hz,1h),5.31(s,2h).
[0193]
实施例10
[0194][0195]
5-((4-((4-氯苄基)氧基)-4'-甲氧基羰基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物10)的制备
[0196]
合成路线:
[0197][0198]
第一步4-((4-氯苄基)氧基)-4'-甲氧基羰基-[1,1
’‑
联苯基]-3-甲醛d6的制备
[0199]
将实施例1中第二步中的3,5-二氯-4-羟基苯硼酸频哪醇酯替换为4-甲氧羰基苯硼酸,1,4-二氧六环替换为n,n-二甲基甲酰胺,进行反应,可得中间体d6,类白色固体715mg,收率62.6%。
[0200]1h nmr(500mhz,dmso-d6)δ10.46(s,1h),8.08
–
8.02(m,4h),7.84(d,j=8.0hz,2h),7.58(d,j=8.0hz,2h),7.49(d,j=8.0hz,2h),7.44(d,j=8.5hz,1h),5.37(s,2h),3.88(s,3h).
[0201]
第二步5-((4-((4-氯苄基)氧基)-4'-甲氧基羰基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物10)的制备
[0202]
以化合物d6(190mg,0.5mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(72mg,0.5mmol)为原料,采用实施例3中相似操作步骤,得到化合物10,橘黄色固体218mg,收率86.2%。
[0203]1h nmr(500mhz,dmso-d6)δ12.45(s,1h),12.32(s,1h),8.58(s,1h),8.51(d,j=2.0hz,1h),8.03(d,j=8.0hz,2h),7.93(dd,j=9.0hz,j=2.0hz,1h),7.79(d,j=8.0hz,2h),7.51(d,j=8.0hz,2h),7.48(d,j=8.0hz,2h),7.31(d,j=9.0hz,1h),5.32(s,2h),3.87(s,3h).
[0204]
实施例11
[0205][0206]
5-((3'-氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物11)的制备
[0207]
合成路线:
[0208][0209]
第一步3'-氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d7的制备
[0210]
于100ml反应瓶中加入化合物b1(976mg,3mmol)、3-氯-4-羟基苯硼酸(543mg,1.05mmol)、四(三苯基膦)钯(105mg,0.09mmol)、碳酸钠(955mg,9mmol),并用氩气保护,注入1,4-二氧六环(22.5ml)和水(4.5ml),105℃加热6小时。冷却,加入1mol/l盐酸(15ml),用乙酸乙酯(15ml)萃取3次,合并有机层。有机层用饱和食盐水洗1次,用无水硫酸钠干燥,过滤,浓缩,得橘色固体1.352g。用n,n-二甲基甲酰胺(20ml)溶解橘色固体,向其中逐滴加入无水乙醇-水(v:v=1:1)溶液15ml后,析出大量棕黄色固体,减压抽滤,水洗,干燥。得化合物d7,棕黄色固体837mg,收率74.8%。
[0211]1h nmr(400mhz,dmso-d6)δ10.44(s,1h),10.31(s,1h),7.92(dd,j=8.8,2.4hz,1h),7.88(d,j=2.4hz,1h),7.63(d,j=2.4hz,1h),7.57(d,j=8.4hz,2h),7.50
–
7.44(m,3h),7.36(d,j=8.8hz,1h),7.05(d,j=8.8hz,1h),5.34(s,2h).
[0212]
第二步5-((3'-氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物11)的制备
[0213]
以化合物d7(112mg,0.3mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(43mg,0.3mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物11,橘黄色固体218mg,收率86.2%。
[0214]1h nmr(400mhz,dmso-d6)δ12.42(s,1h),12.30(s,1h),10.27(s,1h),8.58(s,1h),8.37(d,j=2.4hz,1h),7.78(dd,j=8.8,2.4hz,1h),7.59(d,j=2.4hz,1h),7.52
–
7.46(m,4h),7.42(dd,j=8.4,2.4hz,1h),7.23(d,j=8.8hz,1h),7.04(d,j=8.4hz,1h),5.29(s,2h).
[0215]
实施例12
[0216][0217]
5-((3'-氟-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物12)的制备
[0218]
合成路线:
[0219][0220]
实验步骤:
[0221]
第一步2-((4-氯苄基)氧基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)苯甲醛c1的制备
[0222]
于100ml反应瓶中加入化合物b1(3.255g,10mmol)、联硼酸频那醇酯(2.66g,10.5mmol)、[1,1'-双(二苯基膦基)二茂铁]二氯化钯(220mg,0.3mmol)、醋酸钾(2.94mg,30mmol),并用氩气保护,注入1,4-二氧六环(40ml),95℃加热6小时。冷却,加入水(100ml),用乙酸乙酯(30ml)萃取两次,合并有机层。有机层用饱和食盐水,用无水硫酸钠干燥,过滤,浓缩,硅胶(200-300目)柱色谱分离,石油醚-乙酸乙酯(v:v=100:5~10)混合液为洗脱剂。得中间体c1,类白色固体3.225g,收率86.5%。
[0223]1h nmr(500mhz,dmso-d6)δ10.41(s,1h),8.03(s,1h),7.90(d,j=8.5hz,1h),7.55(d,j=8.0hz,2h),7.48(d,j=8.0hz,2h),7.32(d,j=8.5hz,1h),5.33(s,2h),1.29(s,12h).
[0224]
第二步3'-氟-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d8的制备
[0225]
于25ml反应瓶中加入化合物c1(820mg,2.2mmol)、4-溴-2-氟苯酚(382mg,2mmol)、四(三苯基膦)钯(35mg,0.06mmol)、碳酸钠(636mg,6mmol),并用氩气保护,注入1,4-二氧六环(12ml)和水(3ml),105℃加热6小时。冷却,加入1mol/l盐酸(30ml),用乙酸乙酯(15ml)萃取4次,合并有机层。有机层用饱和食盐水洗1次,用无水硫酸钠干燥,过滤,浓缩,硅胶(200-300目)柱色谱分离,石油醚-乙酸乙酯(v:v=100:25~75)混合液为洗脱剂。得化合物d8,黄色固体416mg,收率58.3%。
[0226]1h nmr(500mhz,dmso-d6)δ10.44(s,1h),9.99(s,1h),7.93
–
7.88(m,2h),7.57(d,j=8.0hz,2h),7.51
–
7.45(m,3h),7.35(d,j=8.5hz,1h),7.31(d,j=8.5hz,1h),7.03(t,j=9.0hz,1h),5.33(s,2h).
[0227]
第二步5-((3'-氟-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物12)的制备
[0228]
以化合物d8(178mg,0.5mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(72mg,0.5mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物12,红色固体162mg,收率67.2%。
[0229]1h nmr(400mhz,dmso-d6)δ12.43(s,1h),12.30(s,1h),9.95(s,1h),8.59(s,1h),8.39(d,j=2.0hz,1h),7.78(dd,j=8.8,2.0hz,1h),7.52
–
7.46(m,4h),7.42(d,j=
12.8hz,1h),7.28(d,j=8.4hz,1h),7.23(d,j=8.8hz,1h),7.02(t,j=8.4hz,1h),5.29(s,2h).
[0230]
实施例13
[0231][0232]
5-((3',5'-二氟-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物13)的制备
[0233]
合成路线:
[0234][0235]
实验步骤:
[0236]
第一步3',5'-二氟-4-((4-氯苄基)氧基)-4'-羟基-[1,1'-联苯基]-3-甲醛d9的制备
[0237]
将实施例12中第二步中的4-溴-2-氟苯酚替换为4-溴-2,6-二氟苯酚,进行反应,可得中间体d9,类白色固体304mg,收率40.5%。
[0238]1h nmr(400mhz,dmso-d6)δ10.44(s,1h),10.31(s,1h),7.96(dd,j=8.8,2.4hz,1h),7.93(d,j=2.4hz,1h),7.57(d,j=8.4hz,2h),7.48(d,j=8.4hz,2h),7.41
–
7.35(m,3h),5.35(s,2h).
[0239]
第二步5-((3',5'-二氟-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物13)的制备
[0240]
以化合物d9(53mg,0.14mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(20mg,0.14mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物13,橘色固体34mg,收率47.9%。
[0241]1h nmr(400mhz,dmso-d6)δ12.44(s,1h),12.31(s,1h),10.28(s,1h),8.56(s,1h),8.40(d,j=1.6hz,1h),7.83(dd,j=8.8,1.6hz,1h),7.52
–
7.47(m,4h),7.34(d,j=12.8hz,1h),7.24(d,j=8.8hz,1h),5.30(s,2h).
[0242]
实施例14
[0243]
[0244]
5-((3',5'-二氯-4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物14)的制备
[0245]
合成路线:
[0246][0247]
实验步骤:
[0248]
第一步3',5'-二氯-4-((4-氯苄基)氧基)-[1,1'-联苯基]-3-甲醛d10的制备
[0249]
将实施例12中第二步中的4-溴-2-氟苯酚替换为3,5-二氯-1-溴苯,进行反应,可得中间体d10,类白色固体339mg,收率86.5%。
[0250]1h nmr(400mhz,dmso-d6)δ10.44(s,1h),8.06(dd,j=8.8,2.4hz,1h),8.02(d,j=2.4hz,1h),7.75(d,j=1.6hz,2h),7.59
–
7.57(m,3h),7.51
–
7.47(m,2h),7.41(d,j=8.8hz,1h),5.37(s,2h).
[0251]
第二步5-((3',5'-二氯-4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物14)的制备
[0252]
以化合物d10(195mg,0.5mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(71mg,0.5mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物14,橘色固体163mg,收率62.9%。
[0253]1h nmr(400mhz,dmso-d6)δ12.45(s,1h),12.34(s,1h),8.54(s,1h),8.47(d,j=2.4hz,1h),7.93(dd,j=8.8,2.4hz,1h),7.69(d,j=2.0hz,2h),7.57(t,j=2.0hz,1h),7.52
–
7.46(m,4h),7.28(d,j=8.8hz,1h),5.32(s,2h).
[0254]
实施例15
[0255][0256]
5-((4-((4-氯苄基)氧基)-4'-羟基-3',5'-二甲基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物15)的制备
[0257]
合成路线:
[0258][0259]
实验步骤:
[0260]
第一步4-((4-氯苄基)氧基)-4'-羟基-3',5'-二甲基-[1,1'-联苯基]-3-甲醛d11
的制备
[0261]
将实施例12中第二步中的4-溴-2-氟苯酚替换为4-溴-2,6-二甲基苯酚,进行反应,可得中间体d11,类白色固体188mg,收率25.1%。
[0262]1h nmr(400mhz,dmso-d6)δ10.45(s,1h),8.36(d,j=2.4hz,1h),7.88
–
7.86(m,1h),7.57(d,j=8.4hz,2h),7.48(d,j=8.4hz,2h),7.33(d,j=9.2hz,1h),7.22(s,2h),5.32(s,2h),2.22(s,6h).
[0263]
第二步5-((4-((4-氯苄基)氧基)-4'-羟基-3',5'-二甲基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物15)的制备
[0264]
以化合物d11(178mg,0.49mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(70mg,0.49mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物15,砖红色固体193mg,收率79.8%。
[0265]1h nmr(400mhz,dmso-d6)δ12.41(s,1h),12.29(s,1h),8.60(s,1h),8.34(d,j=2.4hz,1h),7.72(dd,j=8.8,2.4hz,1h),7.51
–
7.46(m,4h),7.21(d,j=8.8hz,1h),7.18(s,2h),5.27(s,2h),2.21(s,6h).
[0266]
实施例16
[0267][0268]
5-((4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-亚甲基)-1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物16)的制备
[0269]
以化合物d5(99mg,0.3mmol)和1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(52mg,0.3mmol)为原料,采用实施例3中相似操作步骤,得到化合物16,黄色固体83mg,收率58.0%。
[0270]1h nmr(400mhz,dmso-d6)δ8.67(s,1h),8.35(d,j=2.4hz,1h),7.84(dd,j=8.8,2.4hz,1h),7.66
–
7.62(m,2h),7.51
–
7.44(m,6h),7.37
–
7.33(m,1h),7.29(d,j=8.8hz,1h),5.30(s,2h),3.64(s,3h),3.54(s,3h).
[0271]
实施例17
[0272][0273]
5-((3'-氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物17)的制备
[0274]
以化合物d7(112mg,0.3mmol)和1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(52mg,0.3mmol)为原料,采用实施例3中相似操作步骤,得到化合物17,黄色固体117mg,收
率74.1%。
[0275]1h nmr(400mhz,dmso-d6)δ10.28(s,1h),8.63(s,1h),8.25(d,j=2.4hz,1h),7.78(dd,j=8.8,2.4hz,1h),7.59(d,j=2.4hz,1h),7.51
–
7.44(m,4h),7.42(dd,j=8.8,2.4hz,1h),7.24(d,j=8.8hz,2h),7.05(d,j=8.8hz,1h),5.27(s,2h),3.64(s,3h),3.53(s,3h).
[0276]
实施例18
[0277][0278]
5-((4-((4-氯苄基)氧基)-3'-氟-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物18)的制备
[0279]
以化合物d8(108mg,0.3mmol)和1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(52mg,0.3mmol)为原料,采用实施例3中相似操作步骤,得到化合物18,黄色固体128mg,收率83.7%。
[0280]1h nmr(400mhz,dmso-d6)δ9.96(s,1h),8.64(s,1h),8.27(d,j=2.4hz,1h),7.78(dd,j=8.8,2.4hz,1h),7.50
–
7.45(m,4h),7.41(dd,j=12.4,2.0hz,1h),7.28(dd,j=8.8,2.0hz,1h),7.24(d,j=8.8hz,1h),7.02(t,j=8.8hz,1h),5.27(s,2h),3.64(s,3h),3.54(s,3h).
[0281]
实施例19
[0282][0283]
5-(2-((4-氯苄基)氧基)-5-(噻吩-3-基)亚苄基)-1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物19)的制备
[0284]
合成路线:
[0285][0286]
实验步骤:
[0287]
第一步2-((4-氯苄基)氧基)-5-(噻吩-3-基)苯甲醛d12的制备
[0288]
将实施例1中第二步中的3,5-二氯-4-羟基苯硼酸频哪醇酯替换为3-噻吩硼酸,将四(三苯基膦)钯当量由3%改为10%,进行反应,可得中间体d12,白色固体1.297g,收率
65.7%。
[0289]1h nmr(400mhz,dmso-d6)δ10.45(s,1h),8.02(dd,j=8.8,2.4hz,1h),7.99(d,j=2.4hz,1h),7.88(dd,j=2.8,1.2hz,1h),7.64(dd,j=5.2,2.8hz,1h),7.59
–
7.54(m,3h),7.51
–
7.46(m,2h),7.36(d,j=8.8hz,1h),5.34(s,2h).
[0290]
第二步5-(2-((4-氯苄基)氧基)-5-(噻吩-3-基)亚苄基)-1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物19)的制备
[0291]
以化合物d12(164mg,0.5mmol)和1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(86mg,0.5mmol)为原料,将实施例3中的浓硫酸替换为冰醋酸,采用实施例3中相似操作步骤,得到化合物19,橘红色固体136mg,收率56.2%。
[0292]1h nmr(400mhz,dmso-d6)δ8.63(s,1h),8.35(d,j=2.4hz,1h),7.88(dd,j=8.8,2.4hz,1h),7.75(dd,j=2.8,1.6hz,1h),7.64(dd,j=5.2,2.8hz,1h),7.50
–
7.48(dd,j=5.2,1.6hz,1h),7.48
–
7.45(m,4h),7.24(d,j=8.8hz,1h),5.27(s,2h),3.64(s,3h),3.53(s,3h).
[0293]
实施例20
[0294][0295]
5-(2-((4-氯苄基)氧基)-5-(3-氟吡啶-4-基)亚苄基)-1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物20)的制备
[0296]
合成路线:
[0297][0298]
实验步骤:
[0299]
第一步2-((4-氯苄基)氧基)-5-(3-氟吡啶-4-基)苯甲醛d13的制备
[0300]
将实施例1中第二步中的3,5-二氯-4-羟基苯硼酸频哪醇酯替换为3-氟-4-吡啶硼酸,进行反应,可得中间体d13,白色固体375mg,收率75.6%。
[0301]1h nmr(500mhz,dmso-d6)δ10.46(s,1h),8.66(d,j=2.0hz,1h),8.50(d,j=4.5hz,1h),8.01
–
7.98(m,2h),7.70
–
7.66(m,1h),7.59(d,j=8.5hz,2h),7.50
–
7.47(m,3h),5.39(s,2h).
[0302]
第二步5-(2-((4-氯苄基)氧基)-5-(3-氟吡啶-4-基)亚苄基)-1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物20)的制备
[0303]
以化合物d13(68mg,0.2mmol)和1,3-二甲基-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(34mg,0.2mmol)为原料,将实施例3中的浓硫酸替换为浓盐酸,采用实施例3中相似操作步骤,得到化合物20,黄色固体69mg,收率69.7%。
[0304]1h nmr(500mhz,dmso-d6)δ8.75(s,1h),8.60(s,1h),8.57(d,j=5.0hz,1h),8.33(s,1h),7.89(d,j=9.0hz,1h),7.72
–
7.69(m,1h),7.51
–
7.47(m,4h),7.39(d,j=9.0hz,1h),5.33(s,2h),3.64(s,3h),3.51(s,3h).
[0305]
实施例21
[0306][0307]
5-((3',5'-二氯-4-甲氧基-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物21)的制备
[0308]
合成路线:
[0309][0310]
实验步骤:
[0311]
第一步3',5'-二氯-4-甲氧基-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d14的制备
[0312]
将实施例1中第二步中的化合物b1替换为5-溴-2-甲氧基苯甲醛,进行反应,可得中间体d14,白色固体151mg,收率50.8%。
[0313]1h nmr(400mhz,dmso-d6)δ10.35(s,1h),7.76(dd,j=8.8,2.4hz,1h),7.68(d,j=2.4hz,1h),7.26(s,2h),7.18(d,j=8.8hz,1h),3.91(s,3h).
[0314]
第二步5-((3',5'-二氯-4-甲氧基-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物21)的制备
[0315]
以化合物d14(104mg,0.35mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(50mg,0.35mmol)为原料,采用实施例3中相似操作步骤,得到化合物21,橘色固体43mg,收率29.1%。
[0316]1h nmr(500mhz,dmso-d6)δ12.43(s,1h),12.33(s,1h),10.24(s,1h),8.51(s,1h),8.40(d,j=1.5hz,1h),7.87(dd,j=9.0,1.5hz,1h),7.63(s,2h),7.18(d,j=9.0hz,1h),3.94(s,3h).
[0317]
实施例22
[0318][0319]
5-((3',5'-二氯-4-苄氧基-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二
氢嘧啶-4,6(1h,5h)-二酮(化合物22)的制备
[0320]
合成路线:
[0321][0322]
实验步骤:
[0323]
第一步5-溴-2-(苄氧基)苯甲醛b2的制备
[0324]
将实施例1中第一步中的4-氯苄氯替换为氯化苄,进行反应,可得中间体b2,类白色固体2.546g,收率87.5%。
[0325]1h nmr(500mhz,dmso-d6)δ10.33(s,1h),7.82(dd,j=9.0,2.0hz,1h),7.77(d,j=2.0hz,1h),7.51(d,j=8.0hz,2h),7.44
–
7.34(m,3h),7.32(d,j=9.0hz,1h),5.31(s,2h).
[0326]
第二步3',5'-二氯-4-苄氧基-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d15的制备
[0327]
将实施例1中第二步中的化合物b1替换为化合物b2,进行反应,可得中间体d15,白色固体516mg,收率69.2%。
[0328]1h nmr(400mhz,dmso-d6)δ10.44(s,1h),10.27(s,1h),7.98
–
7.95(m,1h),7.92(t,j=2.8hz,1h),7.68
–
7.66(m,2h),7.55(d,j=1.3hz,1h),7.53(s,1h),7.45
–
7.33(m,4h),5.34(s,2h).
[0329]
第三步5-((3',5'-二氯-4-苄氧基-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物22)的制备
[0330]
以化合物d15(112mg,0.3mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(43mg,0.3mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物22,橘黄色固体79mg,收率52.7%。
[0331]1h nmr(500mhz,dmso-d6)δ12.43(s,1h),12.34(s,1h),10.26(s,1h),8.58(s,1h),8.41(d,j=2.0hz,1h),7.85(dd,j=7.5,2.0hz,1h),7.63(s,2h),7.48(d,j=7.0hz,2h),7.42(t,j=7.0hz,2h),7.35(t,j=7.0hz,1h),7.26(d,j=7.5hz,1h),5.31(s,2h).
[0332]
实施例23
[0333]
[0334]
5-((3',5'-二氯-4-((3,5-二氟苯基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物23)的制备
[0335]
合成路线:
[0336][0337]
实验步骤:
[0338]
第一步5-溴-2-((3,5-二氟苄基)氧基)苯甲醛b3的制备
[0339]
将实施例1中第一步中的4-氯苄氯替换为3,5-二氟苄氯,进行反应,可得中间体b3,类白色固体913mg,收率93.1%。
[0340]1h nmr(400mhz,dmso-d6)δ10.35(s,1h),7.83(dd,j=8.8,2.8hz,1h),7.80(d,j=2.8hz,1h),7.30
–
7.46(m,4h),5.33(s,2h).
[0341]
第二步3',5'-二氯-4-((3,5-二氟苯基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d16的制备
[0342]
将实施例1中第二步中的化合物b1替换为化合物b3,进行反应,可得中间体d16,白色固体118mg,收率57.6%。
[0343]1h nmr(400mhz,dmso-d6)δ10.45(s,1h),10.28(s,1h),7.99(dd,j=8.8,2.4hz,1h),7.96(d,j=2.4hz,1h),7.69(s,2h),7.34(d,j=8.8hz,1h),7.32
–
[0344]
7.19(m,3h),5.38(s,2h).
[0345]
第三步5-((3',5'-二氯-4-((3,5-二氟苯基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物23)的制备
[0346]
以化合物d16(61mg,0.15mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(22mg,0.15mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物23,橘黄色固体43mg,收率53.8%。
[0347]1h nmr(400mhz,dmso-d6)δ12.44(s,1h),12.33(s,1h),10.26(s,1h),8.60(s,1h),8.42(d,j=2.4hz,1h),7.86(dd,j=8.8,2.4hz,1h),7.64(s,2h),7.27
–
7.20(m,4h),5.33(s,2h).
[0348]
实施例24
[0349][0350]
5-((3',5'-二氯-5-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物24)的制备
[0351]
合成路线:
[0352][0353]
实验步骤:
[0354]
第一步3-溴-5-((4-氯苄基)氧基)苯甲醛b4的制备
[0355]
将实施例1中第一步中的化合物a1替换为化合物a2,进行反应,可得中间体b4,类白色固体428mg,收率93.9%。
[0356]1h nmr(500mhz,dmso-d6)δ9.93(s,1h),7.69(s,1h),7.60(s,1h),7.52(s,1h),7.51
–
7.46(m,4h),5.23(s,2h).
[0357]
第二步3',5'-二氯-5-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d17的制备
[0358]
将实施例1中第二步中的化合物b1替换为化合物b4,进行反应,可得中间体d17,白色固体277mg,收率67.9%。
[0359]1h nmr(500mhz,dmso-d6)δ10.41(s,1h),10.03(s,1h),7.87(s,1h),7.83(s,2h),7.68(s,1h),7.53(d,j=8.0hz,2h),7.48(d,j=8.0hz,2h),7.45(s,1h),5.28(s,2h).
[0360]
第三步5-((3',5'-二氯-5-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物24)的制备
[0361]
以化合物d17(54mg,0.13mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(19mg,
0.13mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物24,黄色固体47mg,收率66.2%。
[0362]1h nmr(500mhz,dmso-d6)δ12.48(s,1h),12.37(s,1h),10.36(s,1h),8.34(s,1h),8.05(s,1h),7.87(s,1h),7.79(s,2h),7.50
–
7.54(m,3h),7.48(d,j=8.0hz,2h),5.24(s,2h).
[0363]
实施例25
[0364][0365]
5-((3',5'-二氯-6-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物25)的制备
[0366]
合成路线:
[0367][0368]
实验步骤:
[0369]
第一步3-溴-4-((4-氯苄基)氧基)苯甲醛b4的制备
[0370]
将实施例1中第一步中的化合物a1替换为化合物a3,进行反应,可得中间体b4,类白色固体428mg,收率93.9%。
[0371]1h nmr(400mhz,dmso-d6)δ9.86(s,1h),8.13(d,j=2.0hz,1h),7.93(dd,j=8.4,2.0hz,1h),7.53
–
7.49(m,4h),7.40(d,j=8.4hz,1h),5.35(s,2h).
[0372]
第二步3',5'-二氯-6-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d18的制备
[0373]
将实施例1中第二步中的化合物b1替换为化合物b4,进行反应,可得中间体d18,白色固体277mg,收率67.9%。
[0374]1h nmr(400mhz,dmso-d6)δ10.29(s,1h),9.94(s,1h),7.94(dd,j=8.8hz,j=2.0hz,1h),7.92(s,1h),7.61(s,2h),7.48
–
7.43(m,4h),7.41(d,j=8.8hz,1h),5.28(s,2h).
[0375]
第三步5-((3',5'-二氯-6-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物25)的制备
[0376]
以化合物d18(54mg,0.13mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(19mg,0.13mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物25,黄色固体47mg,收率66.2%。
[0377]1h nmr(400mhz,dmso-d6)δ12.41(s,1h),12.32(s,1h),10.29(s,1h),8.52(d,j=2.0hz,1h),8.37(dd,j=8.8,2.0hz,1h),8.34(s,1h),7.61(s,2h),7.50
–
7.44(m,4h),7.34(d,j=8.8hz,1h),5.30(s,2h).
[0378]
实施例26
[0379][0380]
5-((3',5'-二氯-6-((3,5-二氟苯基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物26)的制备
[0381]
合成路线:
[0382][0383]
实验步骤:
[0384]
第一步3-溴-4-((3,5-二氟苄基)氧基)苯甲醛b5的制备
[0385]
将实施例1中第一步中的化合物a1替换为化合物a3,将4-氯苄氯替换为3,5-二氟
苄氯,进行反应,可得中间体b5,类白色固体961mg,收率98.0%。
[0386]1h nmr(400mhz,dmso-d6)δ9.87(s,1h),8.15(d,j=2.0hz,1h),7.95(dd,j=8.8,2.0hz,1h),7.39(d,j=8.8hz,1h),7.27
–
7.20(m,3h),5.38(s,2h).
[0387]
第二步3',5'-二氯-6-((3,5-二氟苯基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d19的制备
[0388]
将实施例1中第二步中的化合物b1替换为化合物b5,进行反应,可得中间体d19,白色固体90mg,收率22.0%。
[0389]1h nmr(400mhz,dmso-d6)δ10.32(s,1h),9.95(s,1h),7.95(dd,j=8.4hz,j=2.0hz,1h),7.94(s,1h),7.64(s,2h),7.39(d,j=8.4hz,1h),7.24
–
7.14(m,3h),5.30(s,2h).
[0390]
第三步5-((3',5'-二氯-6-((3,5-二氟苯基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物26)的制备
[0391]
以化合物d19(61mg,0.15mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(22mg,0.15mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物26,黄色固体69mg,收率86.3%。
[0392]1h nmr(400mhz,dmso-d6)δ12.41(s,1h),12.33(s,1h),10.31(s,1h),8.52(d,j=2.4hz,1h),8.38(dd,j=8.8,2.4hz,1h),8.35(s,1h),7.64(s,2h),7.32(d,j=8.8hz,1h),7.24
–
7.16(m,3h).
[0393]
实施例27
[0394][0395]
5-((3',5'-二氯-6-(吡啶-4-基甲氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物27)的制备
[0396]
合成路线:
[0397][0398]
实验步骤:
[0399]
第一步3-溴-4-(吡啶-4-基甲氧基)苯甲醛b6的制备
[0400]
于100ml反应瓶中加入化合物a3(1.00g,5mmol)、4-(溴甲基)吡啶氢溴酸盐(1.52g,6mmol)、碳酸铯(3.26g,10mmol),并用氩气保护,注入n,n-二甲基甲酰胺(20ml),65℃加热6小时。冷却,加入水(30ml),用乙酸乙酯(45ml)萃取4次,合并有机层。有机层用饱和食盐水洗1次,用无水硫酸钠干燥,过滤,浓缩,硅胶(200-300目)柱色谱分离,石油醚-乙酸乙酯(v:v=100:65~75)混合液为洗脱剂。得化合物b6,类白色固体454mg,收率31.1%。
[0401]1h nmr(400mhz,dmso-d6)δ9.87(s,1h),8.62(d,j=5.2hz,2h),8.16(d,j=2.0hz,1h),7.94(dd,j=8.4,2.0hz,1h),7.48(d,j=5.2hz,2h),7.38(d,j=8.4hz,1h),5.44(s,2h).
[0402]
第二步3',5'-二氯-6-(吡啶-4-基甲氧基)-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d20的制备
[0403]
将实施例1中第二步中的化合物b1替换为化合物b6,进行反应,可得中间体d20,白色固体91mg,收率24.3%。
[0404]1h nmr(400mhz,dmso-d6)δ10.34(s,1h),9.95(s,1h),8.58(d,j=5.6hz,2h),7.94(s,1h),7.93(dd,j=8.4hz,j=2.0hz,1h),7.65(s,2h),7.40(d,j=5.6hz,2h),7.38(d,j=8.4hz,1h),5.36(s,2h).
[0405]
第三步5-((3',5'-二氯-6-(吡啶-4-基甲氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物27)的制备
[0406]
以化合物d20(44mg,0.12mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(16mg,0.12mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物27,黄色固体34mg,收率45.3%。
[0407]1h nmr(400mhz,dmso-d6)δ12.42(s,1h),12.33(s,1h),10.35(s,1h),8.75(d,j=6.0hz,2h),8.52(d,j=2.4hz,1h),8.36(d,j=8.4hz,j=2.4hz,1h),8.34(s,1h),7.68(d,j=6.0hz,2h),7.67(s,2h),7.30(d,j=8.4hz,1h),5.51(s,2h).
[0408]
实施例28
[0409][0410]
5-((3',5'-二氯-6-(噻吩-3-基甲氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物28)的制备
[0411]
合成路线:
[0412][0413]
实验步骤:
[0414]
第一步3-溴-4-(噻吩-3-基甲氧基)苯甲醛b7的制备
[0415]
于25ml反应瓶中加入化合物a3(1.32g,6mmol)、3-噻吩甲醇(623mg,5.5mmol)、偶氮二甲酸二异丙酯(2.2ml,11mmol)、三苯基膦(2.86g,11mmol),并用氩气保护,注入四氢呋喃(20ml),室温,过夜反应。反应液加入水(30ml),用乙酸乙酯(15ml)萃取3次,合并有机层。有机层用饱和食盐水洗1次,用无水硫酸钠干燥,过滤,浓缩,硅胶(200-300目)柱色谱分离,石油醚-乙酸乙酯(v:v=100:10)混合液为洗脱剂。得化合物b7,类白色固体244mg,收率15.0%。
[0416]1h nmr(400mhz,dmso-d6)δ9.86(s,1h),8.12(d,j=2.0hz,1h),7.92(dd,j=8.4,2.0hz,1h),7.64(dd,j=2.8,1.2hz,1h),7.59(dd,j=4.8,2.8hz,1h),7.44(d,j=8.4hz,1h),7.21(dd,j=4.8,1.2hz,1h),5.33(s,2h).
[0417]
第二步3',5'-二氯-6-(噻吩-3-基甲氧基)-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d21的制备
[0418]
将实施例1中第二步中的化合物b1替换为化合物b7,将四(三苯基膦)钯当量由3%改为10%,进行反应,可得中间体d21,白色固体124mg,收率32.7%。
[0419]1h nmr(400mhz,dmso-d6)δ10.28(s,1h),9.93(s,1h),7.94
–
7.90(m,2h),7.60(s,2h),7.58(dd,j=4.8,2.8hz,1h),7.53(dd,j=2.8,1.2hz,1h),7.43(d,j=8.4hz,1h),7.18(dd,j=4.8,1.2hz,1h),5.28(s,2h).
[0420]
第三步5-((3',5'-二氯-6-(噻吩-3-基甲氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物28)的制备
[0421]
于25ml反应瓶中加入化合物d21(57mg,0.15mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(22mg,0.15mmol),注入无水乙醇(5ml),95℃加热4小时。趁热抽滤,滤饼依次用热水(3ml
×
3)、无水乙醇(3ml
×
3)洗,干燥。得化合物28,橘色固体23mg,收率30.3%。
[0422]1h nmr(400mhz,dmso-d6)δ12.40(s,1h),12.32(s,1h),10.28(s,1h),8.52(d,j=2.0hz,1h),8.37(dd,j=8.8,2.0hz,1h),8.34(s,1h),7.60(s,2h),7.59(dd,j=4.8,2.8hz,1h),7.55(dd,j=2.8,1.2hz,1h),7.37(d,j=8.8hz,1h),7.19(dd,j=4.8,1.2hz,1h),5.30(s,2h).
[0423]
实施例29
[0424][0425]
5-((3',5'-二氯-6-(4',4'-二氟环己基-甲氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物29)的制备
[0426]
合成路线:
[0427][0428]
实验步骤:
[0429]
第一步3-溴-4-(4,4-二氟环己基)甲氧基)苯甲醛b8的制备
[0430]
将实施例28中第一步中的3-噻吩甲醇替换为(4,4-二氟环己基)甲醇,进行反应,可得中间体d22,白色固体234mg,收率17.6%。
[0431]1h nmr(400mhz,dmso-d6)δ9.85(s,1h),8.10(d,j=2.0hz,1h),7.92(dd,j=8.8,2.0hz,1h),7.32(d,j=8.8hz,1h),4.09(d,j=6.0hz,2h),2.11
–
1.78(m,7h),1.46
–
1.36(m,2h).
[0432]
第二步3',5'-二氯-6-(4',4'-二氟环己基-甲氧基)-4'-羟基-[1,1
’‑
联苯基]-3-甲d22的制备
[0433]
将实施例1中第二步中的化合物b1替换为化合物b8,进行反应,可得中间体d22,白色固体206mg,收率82.7%。
[0434]1h nmr(400mhz,dmso-d6)δ10.30(s,1h),9.92(s,1h),7.90(d,j=8.8hz,j=2.0hz,1h),7.89(s,1h),7.57(s,2h),7.31(d,j=8.8hz,1h),4.05(d,j=6.0hz,2h),2.07
–
1.98(m,2h),1.90
–
1.74(m,5h),1.41
–
1.31(m,2h).
[0435]
第三步5-((3',5'-二氯-6-(4',4'-二氟环己基-甲氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物29)的制备
[0436]
以化合物d22(62mg,0.15mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(22mg,0.15mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物29,橘黄色固体53mg,收率65.4%。
[0437]1h nmr(400mhz,dmso-d6)δ12.39(s,1h),12.31(s,1h),10.30(s,1h),8.52(d,j=2.0hz,1h),8.36(dd,j=8.8,2.0hz,1h),8.33(s,1h),7.57(s,2h),7.25(d,j=8.8hz,1h),4.08(d,j=6.0hz,2h),2.05
–
1.99(m,2h),1.91
–
1.74(m,5h),1.42
–
1.32(m,2h).
[0438]
实施例30
[0439][0440]
5-((3',5'-二氯-4-((4-氯苄基)氧基)-4'-三氟甲氧基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物30)的制备
[0441]
合成路线:
[0442][0443]
第一步3',5'-二氯-4-((4-氯苄基)氧基)-4'-三氟甲氧基-[1,1
’‑
联苯基]-3-甲醛d23的制备
[0444]
将实施例1中第二步中的3,5-二氯-4-羟基苯硼酸频哪醇酯替换为4-三氟甲氧基苯硼酸,进行反应,可得中间体d23,类白色固体610mg,收率74.9%。
[0445]1h nmr(400mhz,dmso-d6)δ10.46(s,1h),8.00
–
7.98(m,2h),7.80(d,j=8.8hz,2h),7.58(d,j=8.4hz,2h),7.49(d,j=8.4hz,2h),7.46
–
7.41(m,3h),5.36(s,2h).
[0446]
第二步5-((3',5'-二氯-4-((4-氯苄基)氧基)-4'-三氟甲氧基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物30)的制备
[0447]
以化合物d23(122mg,0.3mmol)和2-硫代二氢嘧啶-4,6(1h,5h)-二酮(43mg,0.3mmol)为原料,采用实施例1中第三步相似操作步骤,得到化合物30,黄色固体99mg,收率
61.9%。
[0448]1h nmr(400mhz,dmso-d6)δ12.45(s,1h),12.31(s,1h),8.59(s,1h),8.45(d,j=2.4hz,1h),7.86(dd,j=8.8,2.4hz,1h),7.75(d,j=8.4hz,2h),7.53
–
7.44(m,6h),7.29(d,j=8.8hz,1h),5.32(s,2h).
[0449]
实施例31
[0450][0451]
5-((3'-羧基-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物31)的制备
[0452]
合成路线:
[0453][0454]
实验步骤:
[0455]
第一步6-溴-2,2-二甲基-4h-苯并[d][1,3]二噁英-4-酮a4的制备
[0456]
于100ml反应瓶中加入5-溴水杨酸(2.57g,11.8mmol)、丙酮(1.37g,23.6mmol)、三氟乙酸(16ml)、三氟乙酸酐(10ml),80℃加热24小时。冷却,浓缩,硅胶(200-300目)柱色谱分离,石油醚-乙酸乙酯(v:v=100:15)混合液为洗脱剂。得化合物a4,类白色固体886mg,收率29.2%。
[0457]1h nmr(500mhz,dmso-d6)δ7.96(d,j=2.0hz,1h),7.87(dd,j=8.5,2.0hz,1h),7.13(d,j=8.5hz,1h),1.71(s,6h).
[0458]
第二步3'-羧基-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-甲醛d24的制备
[0459]
将实施例12中第二步中的4-溴-2-氟苯酚替换为化合物a4,进行反应,可得中间体d24,类白色固体129mg,收率20.3%。
[0460]1h nmr(400mhz,dmso-d6)δ10.46(s,1h),8.07(d,j=2.0hz,1h),8.03
–
7.99(m,2h),7.97(d,j=2.4hz,1h),7.58(d,j=8.4hz,2h),7.49(d,j=8.4hz,2h),7.42(d,j=8.8hz,1h),7.22(d,j=8.4hz,1h),5.36(s,2h),1.73(s,6h).
[0461]
第三步5-((3'-羧基-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-亚甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物31)的制备
[0462]
于25ml反应瓶中加入化合物d23(61mg,0.15mmol)、2-硫代二氢嘧啶-4,6(1h,5h)-二酮(25mg,0.15mmol),注入无水甲醇(4ml),加入浓盐酸(0.5ml),65℃加热3小时。趁热抽滤,滤饼依次用热水(3ml
×
3)、无水甲醇(3ml
×
3)洗,干燥。得化合物31,橘色固体40mg,收率78.4%。
[0463]1h nmr(500mhz,dmso-d6)δ13.75(s,1h),12.42(s,1h),12.31(s,1h),11.30(s,1h),8.59(s,1h),8.40(d,j=1.50hz,1h),8.02(d,j=2.0hz,1h),7.81
–
7.76(m,2h),7.51(d,j=8.5hz,2h),7.48(d,j=8.5hz,2h),7.26(d,j=9.0hz,1h),7.06(d,j=8.5hz,1h),5.30(s,2h).
[0464]
实施例32
[0465][0466]
5-((3',5'-二氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物32)的制备
[0467]
合成路线:
[0468][0469]
实验步骤:
[0470]
5-((3',5'-二氯-4-((4-氯苄基)氧基)-4'-羟基-[1,1
’‑
联苯基]-3-基)-甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物32)的制备
[0471]
于25ml反应瓶中加入化合物2(107mg,0.2mmol)和硼氢化钠(23mg,0.6mmol),注入超干乙醇(5ml),0℃反应4小时。反应液浓缩,加水(5ml),用1mol/l盐酸调至ph=4,用乙酸乙酯(15ml
×
3)萃取3次,合并有机层。有机层用饱和食盐水洗1次,用无水硫酸钠干燥,过滤,浓缩,硅胶(200-300目)柱色谱分离,二氯甲烷-甲醇(v:v=100:7-10)混合液为洗脱剂。得化合物32,类白色固体72mg,收率67.3%。
[0472]1h nmr(400mhz,acetone-d6)δ11.02(s,2h),8.81(s,1h),7.60(d,j=8.4hz,2h),7.56(s,2h),7.53
–
7.46(m,2h),7.42(d,j=8.4hz,2h),7.10(d,j=8.4hz,1h),5.23(s,2h),4.06(t,j=6.0hz,1h),3.53(d,j=6.0hz,2h).
[0473]
实施例33
[0474][0475]
5-((4'-三氟甲基-4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物33)的制备
[0476]
合成路线:
[0477][0478]
实验步骤:
[0479]
5-((4'-三氟甲基-4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物33)的制备
[0480]
于25ml反应瓶中加入化合物7(43mg,0.08mmol)和硼氢化钠(10mg,0.26mmol),注入超干乙醇(5ml),0℃反应4小时。反应液浓缩,加水(5ml),用1mol/l盐酸调至ph=1,用乙酸乙酯(15ml
×
3)萃取3次,合并有机层。有机层用饱和食盐水洗1次,用无水硫酸钠干燥,过滤,浓缩,得淡黄色固体51mg。用二氯甲烷(4ml)和甲醇5滴混合溶液打浆,减压抽滤,滤饼用二氯甲烷(3ml
×
3)洗,干燥。得化合物33,类白色固体23mg,收率53.5%。
[0481]1h nmr(400mhz,acetone-d6)δ11.02(s,2h),7.83(d,j=8.4hz,2h),7.75(d,j=8.4hz,2h),7.63
–
7.58(m,4h),7.43(d,j=8.4hz,2h),7.16(d,j=8.4hz,1h),5.25(s,2h),4.08(t,j=6.0hz,1h),3.55(d,j=6.0hz,2h).
[0482]
实施例34
[0483][0484]
5-((4'-氯-4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物34)的制备
[0485]
合成路线:
[0486][0487]
实验步骤:
[0488]
5-((4'-氯-4-((4-氯苄基)氧基)-[1,1
’‑
联苯基]-3-基)-甲基)-2-硫代二氢嘧啶-4,6(1h,5h)-二酮(化合物34)的制备
[0489]
将实施例33中化合物7替换为化合物8进行反应,可得化合物34,白色固体78mg,收率83.0%。1h nmr(400mhz,dmso-d6)δ12.05(s,2h),7.56
–
7.45(m,8h),7.43(dd,j=8.4,2.0hz,1h),7.12(d,j=2.0hz,1h),7.08(d,j=8.4hz,1h),5.22(s,2h),4.06(t,j=6.0hz,1h),3.54(d,j=6.0hz,2h).
[0490]
生物活性测试
[0491]
结核分枝杆菌蛋白酪氨酸磷酸酶b(mptpb)抑制活性评价方法
[0492]
mptpb抑制率的测定:以对硝基苯基磷酸酯(p-nitrophenyl phosphate,pnpp)为底物,在tris-nacl缓冲液(50mm tris/100mm nacl,ph为7.0)中,使用96孔微孔板测定化合物对mptpb蛋白酶的抑制活性。活性测试在含有1.5μg mptpb和50μm待测试化合物的200μl反应体系中进行,先将反应液在室温下孵化10分钟,然后加入pnpp至其浓度为1.3mm(达到其km值),在37℃温度下,使用infinite 200pro分光光度计(tecan)在405nm处测试其反应5分钟时间的吸光度。其中,不含mptpb蛋白酶的反应液作为空白对照组也通过同样的方法测试其吸光度。
[0493]
mptpb ic
50
的测定:对mptpb抑制率超过70%的化合物进一步测试其ic
50
值,通过两倍稀释法配置不同浓度(1.5625
–
100μm)的化合物,在对应浓度下测试其对mptpb的抑制率,使用软件origin 9拟合化合物浓度和抑制率,并计算ic
50
值。
[0494]
所有测试均进行三次独立平行试验。
[0495]
表1.本发明部分化合物mptpb抑制活性
[0496][0497]
由表1可知,本发明化合物具有较强的mptpb抑制活性,特别是化合物2、化合物5、化合物7、化合物8、化合物14、化合物22、化合物24、化合物25和化合物31具有很强的mptpb抑制活性,ic
50
值在1-6μm之间。
[0498]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。