1.本发明属于生物技术技术领域,具体涉及一种具有高表达能力的棒状杆菌和大肠杆菌双表达载体及其构建方法。
背景技术:
2.质粒系统是最常见的用于工程菌株表达外源蛋白的系统,外源蛋白产量的提高,依赖于质粒系统的构建和优化。例如对表达蛋白的启动子和终止子等蛋白表达元件进行优化,来提高蛋白产量。
3.近几年来,对于蛋白表达元件的开发和应用一直是个很热门的话题。有研究团队,利用同义突变的方式对蛋白n端序列进行突变,建库筛选正向突变体。但同义突变的突变体数量有限,且由于是相同的氨基酸组成,效果并不明显。
技术实现要素:
4.本发明属于生物技术领域,具体为一种具有高表达能力的棒状杆菌和大肠杆菌双表达载体及其构建方法,通过在pxmj19载体的外源基因前端插入50bp 核苷酸序列,并对其中的第40bp-第50bp进行人工突变,成功获得比pxmj19 质粒表达能力高约8.5倍的棒状杆菌/大肠杆菌双用载体pxmj19-1676-5’utr 质粒。利用pxmj19质粒表达egfp(绿色荧光蛋白)时的荧光/od值在100000 左右,而利用pxmj19-1676-5’utr质粒表达egfp时的荧光/od值在850000 左右。同时,本发明还公开了该表达质粒的构建方法。
5.不论是利用基因工程技术来进行医药用蛋白的生产,还是食品级蛋白的生产,其产量是关键性问题的同时,安全问题也是必须要考虑的方面。考虑到基因敲除和基因敲入的难度,所以在外源蛋白的“工厂化”生产的时候,质粒系统的应用显得方便又灵活。将外源的目的基因插入到质粒当中,转化入宿主菌,在宿主菌内扩增,生产目的蛋白,达到最终的商业化价值。而目前最常用的大肠杆菌,为一种革兰氏阴性菌,在生产外源蛋白的同时会有内毒素产生,因此并不是药用蛋白或食品级蛋白的最佳生产“工厂”。但由于质粒在大肠杆菌中转化方便,菌株生长周期较短的特点,所以在质粒构建阶段,大肠杆菌这个宿主是首选。而谷氨酸棒状杆菌作为一种革兰氏阳性菌,其不仅仅可以生产分泌性蛋白,与此同时,其分泌蛋白不会形成包涵体也不会有内毒素的干扰。因此将构建好的质粒系统转化入谷氨酸棒状杆菌中进行蛋白生产,有诸多优势。
6.而pxmj19质粒是一种双表达的穿梭质粒,即它既可以在大肠杆菌中完成构建,又能在谷氨酸棒状杆菌中表达。
7.本发明就是在pxmj19穿梭质粒的基础上,通过添加一段谷氨酸棒状杆菌内源基因的5’utr序列,并对此序列进行人为的突变,最终得到可以高效表达的蛋白表达系统。
8.本部分的目的在于概述本发明的实施例的一些方面以及简要介绍一些较佳实施例。在本部分以及本技术的说明书摘要和发明名称中可能会做些简化或省略以避免使本部
分、说明书摘要和发明名称的目的模糊,而这种简化或省略不能用于限制本发明的范围。
9.鉴于上述及现有技术中存在的问题,提出了本发明。
10.因此,本发明的目的在于提供一种具有高表达能力的棒状杆菌和大肠杆菌双表达载体及其构建方法。
11.为解决上述技术问题,根据本发明的一个方面,本发明提供了如下技术方案:一种具有高表达能力的棒状杆菌和大肠杆菌双表达载体及,包括,
12.以pxmj19质粒载体的基因序列为出发序列,在质粒的启动子后端插入一段改造的谷氨酸棒状杆菌内源基因表达启动子的5’utr序列片段,用于外源基因的表达,得到pxmj19-1676-5’utr-外源基因质粒;
13.将pxmj19-1676-5’utr-外源基因质粒转入bzh001或大肠杆菌感受态中表达,即为高表达能力的棒状杆菌和大肠杆菌双表达载体。
14.作为本发明所述高表达能力的棒状杆菌和大肠杆菌双表达载体的一种优选方案,其中:所述pxmj19质粒,核苷酸序列如seqidno.23所示。
15.作为本发明所述高表达能力的棒状杆菌和大肠杆菌双表达载体的一种优选方案,其中:还包括,
16.在pxmj19组成型空载质粒基础上,在启动子序列后端插入一段50bp长度的改造5’utr片段,再接入一段长度为15bp的核苷酸序列,再接入作为表达的外源基因序列,得到pxmj19-1676-5’utr-外源基因质粒;
17.其中,所述外源基因包括绿色荧光蛋白基因片段和红色荧光蛋白基因片段。
18.作为本发明所述高表达能力的棒状杆菌和大肠杆菌双表达载体的一种优选方案,其中:还包括,
19.所述改造5’utr片段,长度为50bp,以seqidno.24为出发序列,对第40bp-第50bp进行人工突变,第40bp-第50bp突变后的核苷酸序列为片段1-4中的一种:
20.片段1的核苷酸序列:cacctcacac;
21.片段2的核苷酸序列:ccgaccacca;
22.片段3的核苷酸序列:gtaggttata;
23.片段4的核苷酸序列:gtccggtatc;
24.所述长度为15bp的核苷酸序列为:aaaggaggacaacta。
25.作为本发明所述高表达能力的棒状杆菌和大肠杆菌双表达载体的构建方法的一种优选方案,其特征在于:包括,
26.获得质粒载体:将pxmj19质粒载体切开;
27.获得改造的外源基因片段:设计上下游引物,上下游引物序列分别如seqidno.2~7、seqidno.8~12所示,对外源基因模板进行扩增反应;得到外源基因片段,进行酶切,得到酶切后的外源基因片段产物,产物进行柱回收;
28.连接:将外源基因片段产物和切开的pxmj19质粒载体连接成pxmj1919-1676-5’utr-外源基因质粒;
29.培养:将pxmj19-1676-5’utr外源基因质粒转化入大肠杆菌或bzh001感受态中表达;
30.其中,由上游引物seqidno.3,下游引物seqidno.2,得到1676-5’utr-egfp片
段1,序列如seqidno.19所示;由上游引物seqidno.4,下游引物seqidno.2,得到1676-5’utr-egfp片段2,序列如seqidno.20所示;由上游引物seqidno.5,下游引物seqidno.2,得到1676-5’utr-egfp片段3,序列如seqidno.21所示;由上游引物seqidno.6,下游引物seqidno.2,得到1676-5’utr-egfp片段4,序列如seqidno.22所示;
31.其中,由上游引物seqidno.8,下游引物seqidno.12,得到1676-5’utr-m-cherry片段1,序列如seqidno.14所示;由上游引物seqidno.9,下游引物seqidno.12,得到1676-5’utr-m-cherry片段2,序列如seqidno.15所示;由上游引物seqidno.10,下游引物seqidno.12,得到1676-5’utr-m-cherry片段3,序列如seqidno.16所示;由上游引物seqidno.11,下游引物seqidno.12,得到1676-5’utr-m-cherry片段4,序列如seqidno.17所示。
32.作为本发明所述高表达能力的棒状杆菌和大肠杆菌双表达载体的构建方法的一种优选方案,其中:还包括,
33.获得质粒载体:将pxmj19质粒载体通过双酶切切开,当外源基因为绿色荧光蛋白基因片段时,酶切位点为hindⅲ和ecorⅰ;当外源基因为红色荧光蛋白基因片段时,hindⅲ和bamhⅰ;
34.获得改造的外源基因片段:设计上下游引物,上下游引物序列分别如seqidno.2~7、seqidno.8~12所示,对外源基因模板使用estaq聚合酶进行pcr的扩增反应;得到外源基因片段,进行双酶切,酶切位点为hindⅲ和ecorⅰ或hindⅲ和bamhⅰ,得到酶切后的外源基因片段产物,产物进行柱回收;
35.连接:使用t4dna连接酶将外源基因片段产物和切开的pxmj19质粒载体连接成pxmj19-1676-5’utr-外源基因质粒;
36.培养:将pxmj19-1676-5’utr-外源基因质粒转化入大肠杆菌或bzh001感受态中表达。
37.作为本发明所述高表达能力的棒状杆菌和大肠杆菌双表达载体的构建方法的一种优选方案,其中:在获得质粒载体步骤中,所述双酶切pxmj19质粒载体体系包括,两种快切酶各5ul,载体质量4500-5000ng,greenbuffer10ul,加无核酸酶灭菌水至100ul。
38.作为本发明所述高表达能力的棒状杆菌和大肠杆菌双表达载体的构建方法的一种优选方案,其中:在获得改造的外源基因片段步骤中,
39.所述扩增反应,为使用estaq聚合酶进行pcr扩增,pcr反应条件为:94℃预变性3min,95℃变性30s,56℃退火30s,72℃延伸,变性、退火、延伸进行共35个循环,72℃保温2min;
40.所述双酶切体系包括两种快切酶各3.5-4ul,片段质量3500-4000ng,greenbuffer10ul,加无核酸酶灭菌水至100ul。
41.作为本发明所述高表达能力的棒状杆菌和大肠杆菌双表达载体的构建方法的一种优选方案,其中:所述在连接步骤中,所述连接体系包括,片段:载体=0.3pmol:0.03pmol,buffer2ul,t4dna连接酶1ul,加无核酸酶灭菌水至20ul,连接温度16℃,连接时间1h。
42.作为本发明所述高表达能力的棒状杆菌和大肠杆菌双表达载体的构建方法的一种优选方案,其中:在培养步骤中,将pxmj19-1676-5’utr-外源基因质粒转化入bzh001感受
态后,冰浴10-15mim,1800v电击两次,将质粒与感受态的混合液打入lbhis培养基中,46℃水浴6min,30℃摇床恢复培养 1.5-2.5h,涂布lbhis 氯霉素抗性的固体培养基平板,30℃培养箱培养24h。
43.本发明的有益效果:
44.本发明提供了一种具有高表达能力的棒状杆菌和大肠杆菌双表达载体及其构建方法,目的在于提高外源蛋白的蛋白产量;从蛋白表达元件的突变入手,对目的基因前的一段序列加以突变,在不引入副产物,不对宿主菌株产生额外的影响下,提高蛋白产量;原pxmj19-egfp的荧光表达量在100000左右,现 pxmj19-1676-5’utr-egfp的荧光表达量在850000左右,对于5’utr核苷酸序列的加入,蛋白表达元件的绿色荧光蛋白表达能力有了8.5倍左右的提升;
45.本发明不仅仅对egfp这种绿色荧光蛋白有效果,对m-cherry的表达也有提升效果,说明,本发明中的质粒系统可以尝试应用于其他的外源蛋白表达。
附图说明
46.为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。其中:
47.图1为pxmj19-egfp组成型质粒图谱;
48.图2为pxmj19-1676-5’utr-egfp组成型质粒图谱;
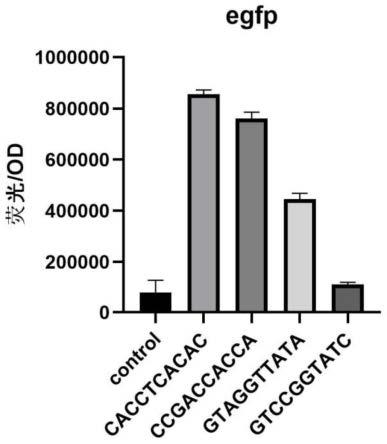
49.图3为egfp(绿色荧光蛋白)在pxmj19-1676-5’utr-egfp组成型质粒系统中的表达验证;其中,从左往右依次为为pxmj19-egfp质粒系统、pxmj19-1676-5’utr-egfp1、2、3、4质粒系统;
50.图4为m-cherry(红色荧光蛋白)在pxmj19-1676-5’utr-m-cherry组成型质粒系统中的表达验证;其中,从左往右依次为为pxmj19-m-cherry质粒系统、 pxmj19-1676-5’utr-m-cherry1、2、3、4质粒系统。
具体实施方式
51.为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例对本发明的具体实施方式做详细的说明。
52.在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
53.其次,此处所称的“一个实施例”或“实施例”是指可包含于本发明至少一个实现方式中的特定特征、结构或特性。在本说明书中不同地方出现的“在一个实施例中”并非均指同一个实施例,也不是单独的或选择性的与其他实施例互相排斥的实施例。
54.本发明实施例中所用化学试剂,若无特殊说明,均为普通市售。
55.本发明实施例所用pxmj19载体质粒序列如seq id no.22所示;快切酶购于takara bio宝日医生物公司,酶切位点为hindⅲ和ecorⅰ或hindⅲ和 bamhⅰ;产物柱购自江苏康为
世纪生物科技股份有限公司,型号为 dnaclean-up kit。
56.实施例中所使用bzh001感受态的制备方法如下:
57.bzh001单菌落在lbb液体培养基中在30℃220r/min的摇床培养12h;12h 后将菌液转接入epo培养基中,并使转接的初始od达到0.3,将转接后的菌液继续在30℃220r/min的摇床培养培养3-5h,使终od达到0.6-0.9;菌液冰浴 25-30min,分装为50ml/管,4500rpm,4℃条件下离心10min;弃上清,加入30ml 预冷过的10%的甘油,将沉淀的菌体重悬,4500rpm,4℃条件下,离心10min,重复三次;最后一次离心后,加入400ul 10%预冷甘油,重悬后,按照80-100ul/ 管的量分装;保存于-80℃冰箱,待使用。
58.实施例1:
59.pxmj19-egfp质粒构建:
60.1.获得质粒载体:应用pxmj19组成型空载质粒通过快切酶双酶切将载体切开,酶切位点为hindⅲ和ecorⅰ。两种快切酶各5ul,载体质量控制在 4500-5000ng之间,green buffer 10ul,最终加无核酸酶灭菌水至100ul。
61.2.获得egfp片段:设计上下游引物,上下游引物序列分别如seq id no.1 和seq id no.2所示,通过引物插入酶切位点;使用estaq聚合酶通过pcr对egfp模板进行反应,pcr反应条件为:94℃预变性3min,95℃变性30s,56℃退火30s,72℃延伸,变性、退火、延伸进行共35个循环,72℃保温2min。
62.得到egfp片段,序列如seq id no.18所示。再应用takara bio宝日医生物公司快切酶,酶切位点为hindⅲ和ecorⅰ,两种快切酶各3.5-4ul,片段质量在3500-4000ng之间,green buffer 10ul,最终加无核酸酶灭菌水至100ul,得到酶切后的egfp片段产物,使用产物进行柱回收。
63.3.连接:用takara bio宝日医生物公司的t4dna连接酶将egfp片段产物和质粒载体连接成pxmj19-egfp质粒,其中,egfp片段产物:载体
64.=0.3pmol:0.03pmol,buffer 2ul,t4dna连接酶为1ul,最终加无核酸酶灭菌水至20ul,在16℃的条件下连接一小时,获得pxmj19-egfp质粒图谱如图1所示。
65.4.培养:将pxmj19-egfp质粒转化入由翊圣生物公司提供的dh5α大肠杆菌感受态中,冰浴25min后,42℃水浴热激45s-1min,热激后再冰浴2-3min,转入37℃摇床进行45-60min的恢复培养。恢复培养后,涂布氯霉素抗性的lbb 培养基平板(lbb培养基:酵母膏5g/l,蛋白胨10g/l,nacl 10g/l,脑心浸出液10g/l,琼脂粉20g/l)。平板培养12-16h,蓝光仪下进行单菌落的初筛选,对有荧光的单菌落进行液体培养基的培养。(液体培养基lbb:酵母膏 5g/l,蛋白胨10g/l,nacl 10g/l,脑心浸出液10g/l)培养12-16h,提取2ml 菌液进行质粒提取,提取纯化后的质粒通过金唯智公司进行质粒序列测序。
66.实施例2:
67.pxmj19-1676-5’utr-egfp质粒构建:
68.pxmj19-1676-5’utr-egfp质粒构建原理和方法与上述pxmj19-egfp质粒构建方法一致,仅步骤2中存在上下游引物的差异,在上游引物中添加 mutation1676序列片段,具体如下。
69.设计上下游引物,上游引物序列分别如seq id no.3~6所示,下游引物序列如seq id no.2所示,通过引物插入酶切位点;使用estaq聚合酶通过pcr 对egfp模板进行反应,
pcr反应条件为:94℃预变性3min,95℃变性30s,56℃退火30s,72℃延伸,变性、退火、延伸进行共35个循环,72℃保温2min。
70.其中,由上游引物seqidno.3,下游引物seqidno.2,得到1676-5’utr-egfp片段1,序列如seqidno.19所示;由上游引物seqidno.4,下游引物seqidno.2,得到1676-5’utr-egfp片段2,序列如seqidno.20所示;由上游引物seqidno.5,下游引物seqidno.2,得到1676-5’utr-egfp片段3,序列如seqidno.21所示;由上游引物seqidno.6,下游引物seqidno.2,得到1676-5’utr-egfp片段4,序列如seqidno.22所示;
71.经产物住回收、连接后,分别得到pxmj19-1676-5’utr-egfp质粒1、2、3、4。pxmj19-1676-5’utr-egfp质粒图谱如图2所示。
72.实施例3:
73.将实施例1、2制备所得质粒分别转入谷氨酸棒状杆菌bzh001的感受态内培养:
74.分别取pxmj19-egfp质粒,pxmj19-1676-5’utr-egfp质粒1、2、3、4各5ul,bzh001感受态80-100ul,分别将质粒打入bzh001感受态中,冰浴10-15mim,1800v电击两次,将质粒与bzh001感受态的混合液打入lbhis培养基中,46℃水浴6min,30℃摇床恢复培养1.5-2.5h,涂布lbhis 氯霉素抗性的固体培养基平板,30℃培养箱培养24h。
75.lbhis培养基:酵母膏2.5g/l,蛋白胨5g/l,nacl5g/l,脑心浸出液18.5g/l,山梨醇91g/l。
76.实施例4:
77.发酵-荧光定量分析:
78.培养24h后的平板菌,在蓝光仪下初步筛选表达绿色荧光的单菌落,挑单菌落在lbb 氯霉素抗性的液体培养基中培养12h,作为菌落的活化阶段,12h后,将活化的菌液转入新的lbb 氯霉素抗性液体培养基发酵24h,通过高通量荧光检测仪测试发酵后的荧光。实验数值如图3所示。
79.与未加入mutation1676序列的pxmj19-egfp质粒相比,四种pxmj19-1676-5’utr-egfp质粒的荧光量菌有不同程度的提升,未加入mutation1676序列的质粒发酵后的荧光/od的比值在1000000左右,而加入mutation1676序列的质粒发酵后的荧光/od的比值在850000左右,最高的荧光量较未突变前增强了8.5倍。
80.实施例5:
81.pxmj19-m-cherry质粒构建:
82.pxmj19-m-cherry质粒构建原理和方法与上述pxmj19-egfp质粒构建方法一致,仅步骤2中存在上下游引物及酶切位点的差异,具体如下。
83.获得m-cherry片段:设计上下游引物,上下游引物序列分别如seqidno.7和seqidno.12所示,通过引物插入酶切位点hindⅲ和bamhⅰ;使用estaq聚合酶通过pcr对m-cherry模板进行反应,pcr反应条件为:94℃预变性3min,95℃变性30s,56℃退火30s,72℃延伸,变性、退火、延伸进行共35个循环,72℃保温2min。
84.得到m-cherry片段,序列如seqidno.13所示。再应用takarabio宝日医生物公司快切酶,酶切位点为hindⅲ和bamhⅰ,两种快切酶各3.5-4ul,片段质量在3500-4000ng之间,greenbuffer10ul,最终加无核酸酶灭菌水至100ul,得到酶切后的m-cherry片段产物,使用产物进行柱回收。
85.再经连接培养,获得pxmj19-m-cherry质粒。
86.实施例6:
87.pxmj19-1676-5’utr-m-cherry质粒构建:
88.pxmj19-1676-5’utr-m-cherry质粒构建原理和方法与上述pxmj19-m-cherry质粒构建方法一致,仅步骤2中存在上下游引物的差异,在上游引物中添加mutation1676序列片段,具体如下。
89.设计上下游引物,上游引物序列分别如seqidno.8~11所示,下游引物序列如seqidno.12所示,通过引物插入酶切位点hindⅲ和bamhⅰ;使用estaq聚合酶通过pcr对egfp模板进行反应,pcr反应条件为:94℃预变性3min,95℃变性30s,56℃退火30s,72℃延伸,变性、退火、延伸进行共35个循环,72℃保温2min。
90.其中,由上游引物seqidno.8,下游引物seqidno.12,得到1676-5’utr-m-cherry片段1,序列如seqidno.14所示;由上游引物seqidno.9,下游引物seqidno.12,得到1676-5’utr-m-cherry片段2,序列如seqidno.15所示;由上游引物seqidno.10,下游引物seqidno.12,得到1676-5’utr-m-cherry片段3,序列如seqidno.16所示;由上游引物seqidno.11,下游引物seqidno.12,得到1676-5’utr-m-cherry片段4,序列如seqidno.17所示;
91.经产物住回收、连接后,分别得到pxmj19-1676-5’utr-m-cherry质粒1、2、3、4。
92.实施例7:
93.将实施例5、6制备所得质粒分别转入谷氨酸棒状杆菌bzh001的感受态内培养:
94.分别取pxmj19-m-cherry质粒,pxmj19-1676-5’utr-m-cherry质粒1、2、3、4各5ul,bzh001感受态80-100ul,分别将质粒打入bzh001感受态中,冰浴10-15mim,1800v电击两次,将质粒与bzh001感受态的混合液打入lbhis培养基中,46℃水浴6min,30℃摇床恢复培养1.5-2.5h,涂布lbhis 氯霉素抗性的固体培养基平板,30℃培养箱培养24h。
95.lbhis培养基:酵母膏2.5g/l,蛋白胨5g/l,nacl5g/l,脑心浸出液18.5g/l,山梨醇91g/l。
96.实施例8:
97.发酵-荧光定量分析:
98.恢复培养24h后的平板菌,在蓝光仪下初步筛选表达绿色荧光的单菌落,挑单菌落在lbb 氯霉素抗性的液体培养基中培养12h,作为菌落的活化阶段,12h后,将活化的菌液转入新的lbb 氯霉素抗性液体培养基发酵24h,通过高通量荧光检测仪测试发酵后的荧光。实验数值如图4所示。
99.与未加入mutation1676序列的pxmj19-m-cherry质粒相比,四种pxmj19-1676-5’utr-m-cherry质粒的荧光量菌有不同程度的提升,虽然与实施例1~4相比,没有之前egfp(绿色荧光蛋白)高达8.5倍的荧光量的提升,但也有4.5倍的提升。验证实验在一定程度上表明这个质粒系统的蛋白表达元件有利于蛋白表达产量的提高,并且有一定的鲁棒性,本发明中的质粒系统可以尝试应用于其他的外源蛋白表达。
100.本发明提供了一种具有高表达能力的棒状杆菌和大肠杆菌双表达载体及其构建方法,目的在于提高外源蛋白的蛋白产量;从蛋白表达元件的突变入手,对目的基因前的一段序列加以突变,在不引入副产物,不对宿主菌株产生额外的影响下,提高蛋白产量;原
pxmj19-egfp的荧光表达量在100000左右,现 pxmj19-1676-5’utr-egfp的荧光表达量在850000左右,对于5’utr核苷酸序列的加入,蛋白表达元件的绿色荧光蛋白表达能力有了8.5倍左右的提升。
101.本发明不仅仅对egfp这种绿色荧光蛋白有效果,对m-cherry的表达也有提升效果,说明,本发明中的质粒系统可以尝试应用于其他的外源蛋白表达。
102.应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。