一种提高l-2-氨基丁酸合成速率的方法
技术领域
1.本发明属于l-2-氨基丁酸合成技术领域,尤其涉及一种提高l-2-氨基丁酸合成速率的方法。
背景技术:
2.目前,l-2-氨基丁酸作为一种非天然氨基酸,可以抑制神经信息的传递,同时可以提高葡萄糖磷酸酯酶的活性,加速脑细胞代谢,作为抗癫痫和抗结核的药物中间体,具有非常好的药理活性。ω-转氨酶是一种磷酸吡哆醛(plp)依赖性酶,可以不对称合成手性胺。
3.oata是一种来源于苍白杆菌(ochrobactrum anthropi)的ω-转氨酶,该酶可以利用α-丁酮酸和异丙胺作为底物,合成l-2-氨基丁酸。然而oata的催化效率较低,难以适应工业生产要求,现如今ω-转氨酶的研究十分火热,通过对其分子进行改造以提高酶活是一种重要的解决方法。
4.通过上述分析,现有技术存在的问题及缺陷为:l-2-氨基丁酸生产方法包括化学合成法和生物合成法,化学合成法价格高昂同时容易造成环境污染,不利于工业应用,相比之下,生物合成法绿色无污染,更适合l-2-氨基丁酸的绿色合成。现有oata的催化效率较低,难以适应工业生产要求。
5.解决以上问题的难度:酶的定向进化具有盲目性,经过多轮长期筛选不一定能选出活性提高的突变体。
6.解决以上问题及缺陷的意义为:筛选出活性提高的突变体,对于工业生产能节约很大成本。
技术实现要素:
7.针对现有技术存在的问题,本发明提供了一种提高l-2-氨基丁酸合成速率的方法,尤其涉及一种基于ω-转氨酶的改造以提高l-2-氨基丁酸的合成速率的方法。
8.本发明是这样实现的,一种提高l-2-氨基丁酸合成速率的方法,所述提高l-2-氨基丁酸合成速率的方法包括以下步骤:
9.步骤一,以α-丁酮酸为底物,通过分子对接预测可突变位点,进行td转化液的配置;
10.步骤二,构建突变体;通过tlc检测筛选出活性高于野生型的突变体;
11.步骤三,对活性高于野生型的突变体进行表达纯化,以纯酶测酶活,进行突变体酶活初筛;
12.步骤四,进行oata野生型及突变体酶活比较、oata组合突变酶活比较。进一步,步骤一中,所述通过分子对接技术进行td转化液的配置,包括:
13.(1)以α-丁酮酸为底物,通过分子对接技术,在参与oata与底物相互作用的氨基酸残基中,选择6个相关位点进行饱和突变;
14.(2)用50mm的磷酸钾缓冲液配置含300mm苏氨酸和450mm异丙胺的底物,用6m hcl
调ph到7.5;
15.(3)用50mm的磷酸钾定容到500ml,向其中加入5g/l的苏氨酸脱氨酶td的发酵菌体,于37℃,220rpm摇床反应4h;
16.(4)反应完后,用tlc进行快速检测,底物苏氨酸反应完全,再用10000rpm离心25min,收集反应上清,即得到td转化液,4℃保存待用。
17.进一步,所述6个相关位点为:y20,l57,w58,g229,a230,m419。
18.进一步,步骤二中,所述突变体的构建方法,包括:
19.(1)以pet28a-oata质粒为模板,通过设计引物对载体进行反扩,对反扩的载体进行dpn i处理;
20.(2)进行t5外切酶介导的同源重组,重组片段转化dh5α感受态,涂布含50μg/ml kana的lb平板,于37℃培养箱培养12-14h;
21.(3)每个平板挑取两个单菌落接种到700μl的含50μg/ml kana的lb液体培养基,37℃,220rpm培养5-6h后送生工进行测序。
22.进一步,步骤二中,所述通过tlc检测筛选出活性高于野生型的突变体,包括:
23.(1)将构建好的突变体及野生型质粒转入bl21,待长出单菌落后,接入到5ml含50μg/ml kana的lb液体培养基中,37℃,220rpm培养;
24.(2)待od600到0.6-0.8时,加入终浓度1mm的iptg,于18℃诱导12h;
25.(3)诱导后,调整野生型及突变体的od600到3,用配置好的td转化液为底物,200μl的体系,于37℃,220rpm反应2h;
26.(4)通过tlc进行快速检测,筛选出活性高于野生型的突变体。
27.进一步,所述层析液的配置方法,包括:
28.1)甲液配置:4g茚三酮溶于400ml正丁醇中,在常温用磁力搅拌器溶解;
29.2)乙液配置:100ml冰醋酸和200ml蒸馏水混合,甲乙液分开,于4℃保存;使用时,甲液:乙液=4:3。
30.进一步,步骤三中,所述对活性高于野生型的突变体进行表达纯化,包括:
31.(1)将单菌落接到5ml的含50μg/ml kana的lb液体培养基中,于37℃,220rpm摇床培养,待od600长到0.6-0.8后,取3ml转接到200ml的含50μg/ml kana的lb液体培养基中,37℃,220rpm摇床培养4-5小时,od600达到0.6-0.8;
32.(2)加终浓度1mm的iptg,18℃诱导12h,随后在8000rpm,10min离心收集菌体,用lysis buffer洗涤菌体两次,再用25ml lysis buffer重悬菌体,设置超声波破碎仪功率为400w,开2.5s,停3.8s,破菌15min,待破菌液澄清透明后,10000rpm离心20min;
33.(3)配置100mm,ph7.4磷酸钠缓冲液,其中取27.72g na2hpo4·
12h2o和3.526g nah2po4·
2h2o,加去离子水定容到1l,配置elution buffer,用elution buffer分别配置10mm,20mm,50mm,200mm的咪唑,向蛋白纯化柱中加入700μl镍珠,用10mm的咪唑预处理,将破菌上清加入到纯化柱中,于4℃静音混合器中混合1h,分别用10mm,20mm咪唑洗三次,再用50mm的咪唑洗一遍,最后用200mm的咪唑洗脱收集;
34.(4)用storage buffer于超滤管中进行除盐。配置1m的底物浓度,5g 95%的α-丁酮酸,6ml异丙胺,溶于50mm的磷酸钾缓冲液中,ph 7.0,用hcl调ph到7.5,再用50mm的磷酸钾定容到46.53ml。以300mm的底物浓度0.25mg/ml的酶量,于37℃反应30min,通过hplc检
测。
35.进一步,所述lysis buffer包括:50mm tris-hcl,50mm nacl,1mmβ-巯基乙醇,0.1mm pmsf,0.5mm plp;所述elution buffer包括:0.5m nacl,0.5mm plp,溶于20mm的磷酸钠缓冲液;所述storage buffer包括:0.15m nacl,0.5mm plp溶于50mm的磷酸钠缓冲液。
36.进一步,步骤三中,所述突变体酶活初筛,包括:
37.(1)oata的催化反应:
38.将oata野生型及突变体的酶浓度调整为2.5mg/ml,将底物用50mm的磷酸钾缓冲液稀释到333.33mm,反应体系:100μl中,加90μl的333.33mm丁酮酸底物,10μl 2.5mg/ml的oata,使得oata的终浓度为0.25mg/ml,α-丁酮酸终浓度为300mm,反应条件:37℃,水浴反应30min;
39.(2)反应的终止:反应30min后,向反应体系中加入100μl的乙腈,涡旋仪振荡30min,使酶变性,12000rpm离心1min;
40.(3)制样:取50μl反应终止液,加450μl的超纯水,混匀,用0.22μm的滤膜过滤,进行液相检测。
41.进一步,步骤四中,所述进行oata野生型及突变体酶活比较、oata组合突变酶活比较,包括:
42.(1)oata野生型及突变体的酶活比较:
43.高效液相色谱检测可以检测出μg级的化合物,野生型及突变体在相同反应条件下,通过检测l-2-氨基丁酸的浓度,来间接比较不同oata的酶活;将每个位点酶活最高的突变体进行双位点组合突变,载体通过单点突变进行反扩,接着进行表达纯化,酶活测定。
44.结合上述所有的技术方案,本发明所具备的优点及积极效果为:本发明提供的提高l-2-氨基丁酸合成速率的方法,基于半理性设计,通过分子对接选取oata活性位点周围的六个位点,通过单点饱和突变及组合突变,筛选出活性提高3.2倍的突变体l57c/m419i,为oata的应用提供强有力的支持。
45.在单点突变中,20位酪氨酸突变为苯丙氨酸及色氨酸后,活性变化不明显,而其他氨基酸的突变会使酶的活性丧失(结合薄层图分析),而然这三个氨基酸都是含有苯环结构的大侧链氨基酸,说明苯环分子的存在有利于酶活性的维持;57位异亮氨酸突变为丙氨酸及半胱氨酸后,活性分别提高了1.6倍和2.7倍,丙氨酸及半胱氨酸均比异亮氨酸小,本发明分析可能突变后,底物更容易进入,而半胱氨酸的巯基于底物形成氢键,有利于底物的稳定;本发明将57c,230s,419i三位点进行组合突变,得到了活性最高突变体l57c/m419i,酶活较野生型提高了3.2倍。
附图说明
46.为了更清楚地说明本发明实施例的技术方案,下面将对本发明实施例中所需要使用的附图做简单的介绍,显而易见地,下面所描述的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下还可以根据这些附图获得其他的附图。
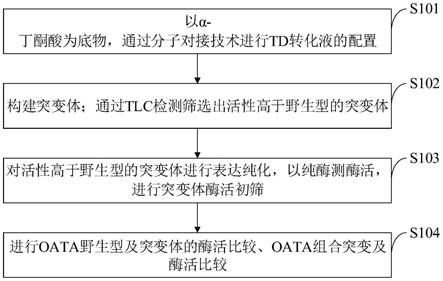
47.图1是本发明实施例提供的提高l-2-氨基丁酸合成速率的方法流程图。
48.图2是本发明实施例提供的y20位点酶活筛选示意图。
49.图3是本发明实施例提供的l57位点酶活筛选示意图。
50.图4是本发明实施例提供的w58位点酶活筛选示意图。
51.图5是本发明实施例提供的g229位点酶活筛选示意图。
52.图6是本发明实施例提供的a230位点酶活筛选示意图。
53.图7是本发明实施例提供的m419位点酶活筛选示意图。
54.图8是本发明实施例提供的单点突变及双点突变酶活示意图。
55.图9是本发明实施例提供的pet28a-oata质粒图谱。
56.具体实施方式如下
57.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
58.本发明实施例的苏氨酸脱氨酶td的发酵菌体,菌体来源于大肠杆菌表达菌株bl21(de3),该菌株为表达常用菌株。
59.针对现有技术存在的问题,本发明提供了一种提高l-2-氨基丁酸合成速率的方法,下面结合附图对本发明作详细的描述。
60.如图1所示,本发明实施例提供的提高l-2-氨基丁酸合成速率的方法包括以下步骤:
61.s101,以α-丁酮酸为底物,通过分子对接预测可突变位点,进行td转化液的配置;
62.s102,构建突变体;通过tlc检测筛选出活性高于野生型的突变体;
63.s103,对活性高于野生型的突变体进行表达纯化,以纯酶测酶活,进行突变体酶活初筛;
64.s104,进行oata野生型及突变体的酶活比较、oata组合突变及酶活比较。
65.下面结合实施例对本发明作进一步描述。
66.1、td转化液配置
67.本发明的oata是一种来源于苍白杆菌(ochrobactrum anthropi)的ω-转氨酶,以α-丁酮酸为底物,通过分子对接技术,研究了参与oata与底物相互作用的氨基酸残基,选择了y20,l57,w58,g229,a230,m419等6个相关位点进行饱和突变,用50mm的磷酸钾缓冲液配置含300mm苏氨酸和450mm异丙胺的底物,用6m hcl调ph到7.5,再用50mm的磷酸钾定容到500ml,向其中加入5g/l的苏氨酸脱氨酶(td)的发酵菌体,于37℃,220rpm摇床反应4h,反应完后,用tlc进行快速检测,底物苏氨酸反应完全,再用10000rpm离心25min,收集反应上清,即得到td转化液,4℃保存待用。
68.2、突变体筛选
69.2.1突变体的构建
70.以pet28a-oata质粒为模板,通过设计引物对载体进行反扩,对反扩的载体进行dpn i处理,然后进行t5外切酶介导的同源重组,重组片段转化dh5α感受态,涂布含50μg/ml kana的lb平板,于37℃培养箱培养12-14h,每个平板挑取两个单菌落接种到700μl的含50μg/ml kana的lb液体培养基,37℃,220rpm培养5-6h后送生工进行测序。
71.将构建好的突变体及野生型质粒转入bl21(de3),待长出单菌落后,接入到5ml含50μg/ml kana的lb液体培养基中,37℃,220rpm培养,待od600到0.6-0.8时,加入终浓度1mm
的iptg,于18℃诱导12h,诱导后,调整野生型及突变体的od600到3,用配置好的td转化液为底物,200μl的体系,于37℃,220rpm反应2h,通过tlc进行快速检测,筛选出活性高于野生型的突变体。层析液的配置:甲液:4g茚三酮溶于400ml正丁醇中,在常温用磁力搅拌器溶解;乙液:100ml冰醋酸和200ml蒸馏水混合,甲乙液分开,于4℃保存。使用时甲液:乙液=4:3,通过初筛,发现l57c,m419i具有比较好的活性。
72.对活性高于野生型的突变体进行表达纯化,以纯酶测酶活。将单菌落接到5ml的含50μg/ml kana的lb液体培养基中,于37℃,220rpm摇床培养,待od600长到0.6-0.8后,取3ml转接到200ml的含50μg/ml kana的lb液体培养基中,37℃,220rpm摇床培养4-5小时,od600达到0.6-0.8,然后加终浓度1mm的iptg,18℃诱导12h,随后在8000rpm,10min离心收集菌体,用lysis buffer(50mm tris-hcl,50mm nacl,1mmβ-巯基乙醇,0.1mm pmsf,0.5mm plp)洗涤菌体两次,再用25ml lysis buffer重悬菌体,设置超声波破碎仪功率为400w,开2.5s,停3.8s,破菌15min,待破菌液澄清透明后,10000rpm离心20min。配置100mm,ph7.4磷酸钠缓冲液,其中取27.72g na2hpo4·
12h2o和3.526g nah2po4·
2h2o,加去离子水定容到1l,配置elution buffer(0.5m nacl,0.5mm plp,溶于20mm的磷酸钠缓冲液),用elution buffer分别配置10mm,20mm,50mm,200mm的咪唑,向蛋白纯化柱中加入700μl镍珠,用10mm的咪唑预处理,将破菌上清加入到纯化柱中,于4℃静音混合器中混合1h,分别用10mm,20mm咪唑洗三次,再用50mm的咪唑洗一遍,最后用200mm的咪唑洗脱收集。用storage buffer(0.15m nacl,0.5mm plp溶于50mm的磷酸钠缓冲液)于超滤管中进行除盐。配置1m的底物浓度,5gα-丁酮酸(麦克林公司,纯度:95%,cas:600-18-0),6ml异丙胺,溶于50mm的磷酸钾缓冲液中(ph 7.0),用hcl调ph到7.5,再用50mm的磷酸钾定容到46.53ml。以300mm的底物浓度0.25mg/ml的酶量,于37℃反应30min,通过hplc检测,其中l57c酶活较野生型提高了2.7倍,m419i较野生型提高了1.8倍。将这两个位点的氨基酸进行组合突变,突变体l57c/m419i活性提高了3.2倍。
73.本发明通过计算机模拟对预测出的六个位点进行饱和突变和组合突变,筛选出了对α-丁酮酸催化活性提高3.2倍的突变体,具有非常好的应用前景。
74.2.2突变体酶活初筛
75.y20位点酶活筛选如图2所示,l57位点酶活筛选如图3所示,w58位点酶活筛选如图4所示,g229位点酶活筛选如图5所示,a230位点酶活筛选如图6所示,m419位点酶活筛选如图7所示。
76.本发明通过薄层层析显色的斑点大小和颜色来比较突变体及野生型的酶活,这是一种半定量分析,随后本发明对活性提高的部分突变体进行表达纯化,对反应产物进行液相检测,及定量分析oata突变体活性提高倍数。
77.2.2.1oata的催化反应
78.将oata野生型及突变体的酶浓度调整为2.5mg/ml,将底物用50mm的磷酸钾缓冲液稀释到333.33mm,反应体系:100μl中,加90μl的333.33mm丁酮酸底物,10μl 2.5mg/ml的oata,使得oata的终浓度为0.25mg/ml,α-丁酮酸终浓度为300mm,反应条件:37℃,水浴反应30min。
79.2.2.2反应的终止
80.反应30min后,向反应体系中加入100μl的乙腈,涡旋仪振荡30min,使酶变性,
12000rpm离心1min。
81.2.2.3制样
82.取50μl反应终止液,加450μl的超纯水,混匀,用0.22μm的滤膜过滤,进行液相检测。
83.3、oata野生型及突变体的酶活比较
84.高效液相色谱检测可以检测出μg级的化合物,野生型及突变体在相同反应条件下,本发明通过检测l-2-氨基丁酸的浓度,来间接比较不同oata的酶活。
85.4、oata组合突变及酶活比较
86.本发明将每个位点酶活最高的突变体进行双位点组合突变,载体通过单点突变进行反扩,接着进行表达纯化,酶活测定,方法同上,单点突变及双点突变酶活如图8所示。
87.在单点突变中,20位酪氨酸突变为苯丙氨酸及色氨酸后,活性变化不明显,而其他氨基酸的突变会使酶的活性丧失(结合薄层图分析),而然这三个氨基酸都是含有苯环结构的大侧链氨基酸,说明苯环分子的存在有利于酶活性的维持;57位异亮氨酸突变为丙氨酸及半胱氨酸后,活性分别提高了1.6倍和2.7倍,丙氨酸及半胱氨酸均比异亮氨酸小,本发明分析可能突变后,底物更容易进入,而半胱氨酸的巯基于底物形成氢键,有利于底物的稳定;本发明将57c,230s,419i三位点进行组合突变,得到了活性最高突变体l57c/m419i,酶活较野生型提高了3.2倍。
88.5、意义
89.本发明基于一种半理性设计,通过分子对接选取了oata活性位点周围的六个位点,通过单点饱和突变及组合突变,筛选出了活性提高3.2倍的突变体l57c/m419i,为oata的应用提供了强有力的支持。
90.野生型oata原始dna序列见seq id no:1,oata野生型蛋白质序列见seq id no:2。
91.pet28a-oata质粒图谱如图9所示。
92.本发明实施例基于半理性设计将α-丁酮酸与苍白杆菌来源的ω-转氨酶(简称oata,pdb:5ghf)进行分子对接,其中苍白杆菌来源的ω-转氨酶基因序列来源于ncbi搜索,已公开,在α-丁酮酸与活性位点范围内,选出了六个氨基酸位点进行饱和突变。
93.与野生型中α-丁酮酸的对接模型相比,突变体活性部位α-丁酮酸结合的改变导致底物羰基碳与pmp之间的距离从显著减小到这可能是催化活性提高的原因之一。分子动力学模拟结果表明,在底物接近活性位点的过程中,i419和底物之间的疏水作用被破坏,c57和α-丁酮酸之间形成氢键,导致底物的羰基碳向pmp转移。底物与突变体活性位点之间的可及性和稳定性的提高可能导致kcat的提高。
94.以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。