靶向a
2a
的苯并咪唑并吡嗪-3-甲酰胺及其肿瘤免疫功能
技术领域
1.本发明属于医药技术领域,具体涉及靶向a
2a
的苯并咪唑并吡嗪-3-甲酰胺及其肿瘤免疫功能。
背景技术:
2.基于免疫检查点抑制或过继细胞疗法的癌症免疫疗法彻底改变了癌症的治疗方式,然而,很大一部分患者却并未从此类治疗中受益。在肿瘤微环境(tme)中活跃的多个冗余和非冗余免疫抑制通路可以部分解释当前免疫检查点治疗的低响应率问题。癌症免疫逃避的一个关键机制是在tme内产生高水平的免疫抑制性腺苷。肿瘤微环境中大量存在的腺苷会抑制t细胞、nk细胞等免疫细胞的增殖、成熟及免疫细胞活素的产生,从而导致机体的免疫损伤,并促进肿瘤细胞的增长。不仅如此,腺苷对肿瘤细胞还有直接的增殖作用。而且靶向阻断该类腺苷受体的主要效应物目前已被证明可在各种临床癌症模型中促进抗肿瘤免疫,从而提高标准癌症治疗和其他免疫检查点阻断治疗的疗效。
3.腺苷受体属于g蛋白偶联受体(gpcr),可细分为a
2a
、a
2b
、a1和a3四个亚型,在生理条件下,胞外atp(eatp)和eado水平都保持在纳摩尔范围内。但在缺氧、营养饥饿或炎症引发细胞死亡或细胞应激后,atp会迅速释放到细胞外空间达到微摩尔浓度,此时肿瘤细胞为了适应组织损伤和缺氧的环境,其在缺氧诱导因子hif-1的调控下,会通过cd39和cd73这两个酶将细胞外的三磷酸腺苷atp转化为amp,再转化为腺苷,由此产生大量的胞外腺苷积累在tem中。而高浓度的细胞外腺苷主要与免疫细胞膜表面的a
2a
r相互作用,a
2a
r通过与gs/golf蛋白结合,促进腺苷酸环化酶(ac)活性的增加并增加胞内环磷酸腺苷(camp)的含量,进而通过激活蛋白激酶a(pka)而激活许多离子通道和受体,将免疫抑制信号传递到免疫细胞内,从而抑制t细胞、nk细胞、巨噬细胞和树突状dc细胞等免疫细胞的增殖、成熟、浸润以及免疫细胞因子的产生,最终导致机体的免疫损伤和肿瘤细胞的免疫逃逸。因此,通过设计a
2a
受体拮抗剂药物分子来阻断腺苷a
2a
受体介导的信号传递,从而阻止机体的免疫损失,并抑制肿瘤细胞的增长,是目前处于临床研究阶段的一种新型肿瘤免疫治疗策略。
4.到目前为止,还没有正式被获批上市的腺苷a
2a
受体拮抗剂药物分子,只有7个小分子a
2a
受体拮抗剂处于临床研究阶段,它们分别是astrazeneca公司的azd4635、corvus公司的cpi-444、novartis公司的pbf-509、恒瑞医药的shr5126、基石药业的cs3005、exscientia公司的exs21546以及arcus biosciences公司开发的同时靶向a
2a
和a
2b
的ab928。这7个候选药物分子正处于临床测试黑色素瘤、肺鳞状细胞癌、肠癌、胰腺癌、肾癌、膀胱癌、卵巢癌、前列腺癌等常见肿瘤疾病的研究当中,它们对人体特定肿瘤的具体疗效和毒性数据尚未公布,未来能否成功上市也尚未可知,因而研发新的腺苷a
2a
受体拮抗药物具有重要的科学研究意义和临床医学应用价值。
技术实现要素:
5.为了克服上述现有技术的不足,本发明的目的是提供一种亚型选择性腺苷a
2a
受体
拮抗剂,所述拮抗剂为1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物,本发明经研究发现1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物具有较好的增强肿瘤免疫作用,在制备肿瘤免疫治疗药物中具有广泛的用途。
[0006]
为实现上述发明目的,发明所采用的技术方案为:
[0007]
一种亚型选择性腺苷a
2a
受体拮抗剂,所述拮抗剂为1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物(其分子式为c
17h14
n6o,分子量为318.34),所述拮抗剂的结构如式(ⅰ)所示:
[0008][0009]
本发明还提供了所述的亚型选择性腺苷a
2a
受体拮抗剂在制备靶向腺苷a
2a
受体的药物中的应用。
[0010]
经研究发现,本发明的1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物可特异性靶向a
2a
r。
[0011]
本发明还提供了所述的亚型选择性腺苷a
2a
受体拮抗剂在制备肿瘤免疫治疗药物中的应用。
[0012]
本发明研究发现,特定结构的1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物具有较好的增强肿瘤免疫的作用,在制备肿瘤免疫治疗药物中具有广泛的用途。
[0013]
优选地,所述肿瘤包括但不限于乳腺癌。
[0014]
优选地,所述免疫治疗为抑制camp的累积。
[0015]
优选地,所述免疫治疗为促进免疫细胞分泌细胞因子。进一步地,所述细胞因子为il-2。
[0016]
优选地,所述免疫治疗为增强免疫细胞对肿瘤的杀伤作用。
[0017]
经研究发现,本发明的1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物可特异性靶向a
2a
r,抑制camp累积、促进免疫细胞细胞因子释放,增强共培养中免疫细胞对肿瘤细胞的杀伤,增强肿瘤免疫治疗效果。目前肿瘤免疫治疗响应率低,免疫逃逸频发,本发明为靶向a
2a
r增强免疫治疗的新药开发提供了重要参考,且为减少免疫逃逸提供了一定的治疗策略,具有良好的应用前景。
[0018]
本发明还提供了所述的亚型选择性腺苷a
2a
受体拮抗剂在制备抑制肿瘤细胞生长的药物中的应用。
[0019]
优选地,所述肿瘤细胞包括但不限于乳腺癌细胞。
[0020]
通过balb/c小鼠皮下细胞移植瘤模型实验发现,1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物能够抑制乳腺癌细胞的生长。
[0021]
本发明还提供了一种靶向腺苷a
2a
受体的药物或一种肿瘤免疫治疗药物或一种抑制肿瘤细胞生长的药物,所述药物以所述的亚型选择性腺苷a
2a
受体拮抗剂作为主要活性成分。
[0022]
优选地,在上述应用及药物方案中,所述的亚型选择性腺苷a
2a
受体拮抗剂还包括1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物在药学上可以接受的盐或溶剂合物。
[0023]
术语“可接受的盐”是指上述化合物或其立体异构体与无机和/或有机酸和碱形成的酸式和/或碱式盐,也包括两性离子盐(内盐),还包括季铵盐,例如烷基铵盐。这些盐可以是在化合物的最后分离和纯化中直接得到。也可以是通过将上述化合物,或其立体异构体,与一定数量的酸或碱适当(例如等当量)进行混合而得到。这些盐可能在溶液中形成沉淀而以过滤方法收集,或在溶剂蒸发后回收而得到,或在水介质中反应后冷冻干燥制得。
[0024]
更优选地,所述药学上可以接受的盐为药学上可以接受的无机盐或有机盐。
[0025]
进一步地,药学上可以接受的盐包括但不限于:硫酸盐、柠檬酸盐、乙酸盐、草酸盐、氯化物、溴化物、碘化物、硝酸盐、硫酸氢盐、磷酸盐、酸式磷酸盐、异烟酸盐、乳酸盐、水杨酸盐、酸式柠檬酸盐、酒石酸盐、油酸盐、鞣酸盐、泛酸盐、酒石酸氢盐、抗坏血酸盐、琥珀酸盐、马来酸盐、龙胆酸盐、富马酸盐、葡糖酸盐、葡糖醛酸盐、糖酸盐、甲酸盐、苯甲酸盐、谷氨酸盐,甲烷磺酸盐(甲磺酸盐)、乙烷磺酸盐、苯磺酸盐、对甲苯磺酸盐、和双羟萘酸盐;或者铵盐(例如伯胺盐、仲胺盐、叔胺盐、季铵盐)、金属盐(例如钠盐、钾盐、钙盐、镁盐、锰盐、铁盐、锌盐、铜盐、锂盐、铝盐)。
[0026]
优选地,在上述药物方案中,还包括一种或多种药学上可接受的载体、稀释剂或赋形剂。
[0027]
术语“药学上可接受的”是指某载体、稀释剂或赋形剂,和/或所形成的盐通常在化学上或物理上与构成某药物剂型的其它成分相兼容,并在生理上与受体相兼容。
[0028]
优选地,在上述药物方案中,所述药物的剂型包括但不限于注射剂、胶囊剂、片剂、丸剂、颗粒剂。
[0029]
更有选地,优选地,在上述药物方案中,所述药物的剂型为注射剂。
[0030]
与现有技术相比,本发明的有益效果是:
[0031]
本发明公开了一种亚型选择性腺苷a
2a
受体拮抗剂,所述拮抗剂为1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物,分子式为c
17h14
n6o,分子量为318.34。本发明经研究发现,所述的1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物可特异性靶向a
2a
r,抑制camp累积、促进免疫细胞细胞因子释放,增强共培养中免疫细胞对肿瘤细胞的杀伤,增强肿瘤免疫治疗效果。在分子水平、细胞水平和癌症小鼠模型中均表现出明显的肿瘤免疫增强作用,有望应用于肿瘤免疫治疗,在制备肿瘤免疫治疗药物中具有广泛的用途。
附图说明
[0032]
图1为ldh-e-4对a
2a
r的靶向特性和细胞功能测试结果(a为ldh-e-4与a
2a
、a1受体结合的ic
50
曲线;b为ldh-e-4抑制camp累积的ic
50
曲线;c为ldh-e-4促进il-2释放的ec
50
曲线);
[0033]
图2为ldh-e-4体内外增强免疫细胞杀伤肿瘤细胞的效果(a为ldh-e-4增强pbmc对mda-mb-231乳腺癌细胞的杀伤能力;b为在小鼠皮下乳腺癌细胞移植瘤模型中给药期间的肿瘤体积变化情况;c为为给药26天后,剥离的乳腺癌皮下肿瘤的质量图;d为给药26天后,剥离的乳腺癌皮下肿瘤的外观图)。
具体实施方式
[0034]
下面对本发明的具体实施方式作进一步说明。在此需要说明的是,对于这些实施方式的说明用于帮助理解本发明,但并不构成对本发明的限定。此外,下面所描述的本发明各个实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互组合。
[0035]
下述实施例中的实验方法,如无特殊说明,均为常规方法,下述实施例中所用的试验材料,如无特殊说明,均为可通过常规的商业途径购买得到。
[0036]
实施例1 1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺类小分子化合物(ldh-e-4)的制备
[0037]
ldh-e-4的结构如下所示:
[0038][0039]
该化合物的制备包括如下步骤:
[0040]
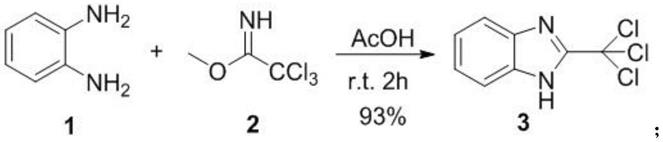
(1)式3所示化合物的制备:
[0041][0042]
根据上述反应式,室温下将式1所示化合物(1,2-苯二胺,3.2g,30mmol),乙酸(acoh)50ml加入到反应瓶中,冰浴冷却后缓缓加入起始原料,即式2所示化合物(2,2,2-三氯亚氨逐乙酸甲酯,4ml,30mmol),加完后室温搅拌2小时,并通过tlc显示反应完成。反应结束后将反应物过滤,所得滤饼经水洗(3
×
25ml)和真空干燥后得到式3所示化合物(2-三氯甲基-苯并嘧啶)(7.1g,93%)。
[0043]
(2)式4所示化合物的制备:
[0044][0045]
根据上述反应式,将式3所示化合物(2-三氯甲基苯并嘧啶,5.9g,25mmol)冷却至0度,然后在0度下加入氨的1,4-二氧六环溶液(0.40m,125ml)并密封,室温下搅拌2小时,通
过tlc显示反应完成后减压浓缩,过柱,得到式4所示化合物(2-氰基苯并嘧啶,2.7g,76%)。
[0046]
(3)式6所示化合物的制备:
[0047][0048]
根据上述反应式,在-15℃下将乙腈mecn(40ml),n,n-二异丙基乙胺dipea(4.4g,34mmol),式4所示化合物(2-氰基苯并嘧啶,2.6g,17mmol)和式5所示化合物(3-溴-2-氧代丙基乙酸酯,3.3g,17mmol)加入到反应瓶中,-15℃度搅拌2小时,通过tlc显示反应完成后加水稀释,随后经乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩、过柱,得到式6所示化合物(1-(2-(4-甲氧苯基)-2-氧杂乙基)-1氢-苯并嘧啶-2-氰)(3.5g,80%)。产物的图谱信息如下:
[0049]1h nmr(400mhz,chloroform-d)δ7.89
–
7.86(m,1h),7.50
–
7.40(m,2h),7.30
–
7.28(m,1h),5.28(s,2h),4.81(s,2h),2.22(s,3h)。
[0050]
(4)式7所示化合物的制备:
[0051][0052]
根据上述反应式,将式6所述化合物(3-(2-氰基-1h-苯并[d]咪唑-1-基)-2-氧代丙基乙酸酯,3.3g,13mmol),醋酸铵nh4oac(5.1g,66mmol)和醋酸hoac(10ml)加入密封管中,95度搅拌1小时,通过tlc显示反应完成后加入饱和碳酸氢钠溶液中和反应至不再产生气泡,使用二氯甲烷萃取(3
×
100ml),收集有机相,有机相经无水硫酸钠干燥、减压浓缩、过柱,得到式7所示化合物(1-氨基苯并[4,5]咪唑并[1,2-a]吡嗪-3-基)乙酸甲酯)(2.8g,85%)。产物的图谱信息如下:
[0053]1h nmr(500mhz,chloroform-d)δ7.96
–
7.86(m,3h),7.56
–
7.46(m,3h),6.06(s,2h),5.10(s,2h),2.16(s,3h)。
[0054]
(5)式8所示化合物的制备:
[0055][0056]
根据上述反应式,将式7所示化合物(1-氨基苯并[4,5]咪唑并[1,2-a]吡嗪-3-基)乙酸甲酯,2.8,11mmol)、4-二甲氨基吡啶dmap(134mg,1.1mmol)、三乙胺tea(4.5g,44mmol)溶于50ml四氢呋喃thf中,并冷却至0度,然后在0度下缓慢滴加二碳酸二叔丁酯(boc)2o(6.0g,27.5mmol),室温搅拌4小时,通过tlc显示反应完成后加水100ml,使用二氯甲烷萃取
(3
×
100ml),合并有机相,有机相经100ml饱和食盐水洗涤、硫酸钠干燥、减压浓缩、过柱,得到式8所示化合物((1-(二(叔丁氧基羰基)氨基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-基)乙酸甲酯,4.2g,84%)。产物的图谱信息如下:
[0057]1h nmr(400mhz,chloroform-d)8.46(s,1h),8.07(d,j=8.3hz,1h),7.97(d,j=8.3hz,1h),7.64(t,j=7.7hz,1h),7.53(t,j=7.7hz,1h),5.29(s,2h),2.16(3h,s),1.41(s,18h)。
[0058]
(6)式9所示化合物的制备:
[0059][0060]
根据上述反应式,将式8所示化合物((1-(二(叔丁氧基羰基)氨基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-基)乙酸甲酯,4.1g,9mmol)溶于50ml甲醇meoh中,冷至0℃后缓慢加入碳酸钾(3.7g,27mmol),室温搅拌4h,并通过tlc显示反应完成,反应后加水100ml,使用二氯甲烷萃取(3
×
100ml),合并有机相,有机相经饱和食盐水洗涤(100ml)、无水硫酸钠干燥、减压浓缩、过柱,得到式9所示化合物((3-(羟甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-1-基)氨基甲酸叔丁酯,67%,1.9g)。产物的图谱信息如下:
[0061]1h nmr(400mhz,chloroform-d)δ8.56(s,1h),8.12(s,1h),7.96(d,j=8.3hz,1h),7.89(d,j=8.3hz,1h),7.61
–
7.57(m,1h),7.50-7.60(m,1h),4.85(s,2h),1.56(s,9h)。
[0062]
(7)式10所示化合物的制备:
[0063][0064]
根据上述反应式,将式9所示化合物((3-(羟甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-1-基)氨基甲酸叔丁酯,1.9g,6mmol)溶于50ml二氯甲烷dcm中,冷至0℃后缓慢加入戴斯马丁氧化剂——戴斯-马丁高碘烷dmp(3.6g,8.4mmol),室温搅拌4h,通过tlc显示反应完成后过滤,加饱和碳酸氢钠水溶液至不再产生气泡,随后使用二氯甲烷萃取(3
×
100ml),合并有机相,有机相经饱和食盐水洗涤(100ml)、无水硫酸钠干燥、减压浓缩、过柱,得到式10所示化合物((3-甲酰基苯并[4,5]咪唑并[1,2-a]吡嗪-1-基)氨基甲酸叔丁酯,1.4g,75%)。产物的图谱信息如下:
[0065]1h nmr(500mhz,chloroform-d)δ10.17(s,1h),8.82(s,1h),8.03
–
8.00(m,2h),7.70
–
7.67(m,1h),7.61
–
7.57(m,1h),1.61(s,9h)。
[0066]
(8)式11所示化合物的制备:
[0067][0068]
根据上述反应式,将式10所示化合物(3-甲酰基苯并[4,5]咪唑并[1,2-a]吡嗪-1-基)氨基甲酸叔丁酯,1.4g,4.5mmol)、磷酸二氢钠(2.2g,18mmol)、异戊烯2-methylbut-2-ene(1.6g,22mmol)溶于40ml四氢呋喃thf和20ml水的混合溶液中,冷至0℃后缓慢加入亚氯酸钠(1.6g,18mmol),0℃搅拌3h,通过tlc显示反应完成后,0℃下加入硫代硫酸钠(3.6g,22mmol)水溶液淬灭反应,随后使用二氯甲烷萃取(3
×
100ml),合并有机相,有机相经饱和食盐水洗涤(100ml)、无水硫酸钠干燥、减压浓缩后所得固体用正己烷洗涤,得到式11所示化合物(1-((叔丁氧基羰基)氨基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-羧酸,886mg,60%)。产物的图谱信息如下:
[0069]1h nmr(400mhz,dmso-d6)δ9.46(s,1h),8.56(s,1h),7.98-7.96(m,1h),7.65
–
7.54(m,2h),1.50(s,9h)。
[0070]
(9)式13所示化合物的制备:
[0071][0072]
根据上述反应式,将式11所示化合物(1-((叔丁氧基羰基)氨基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-羧酸,67mg,0.2mmol)、式12所示化合物(2-氨甲基吡啶,32mg,0.3mmol)、1-羟基苯并三氮唑hobt(41mg,0.3mmol)溶于2ml二氯甲烷dcm中,冷却至0℃后缓慢加入二环己基碳二亚胺dcc(62mg,0.3mmol),室温搅拌16小时,通过tlc显示反应完成后使用硅胶过滤,并用二氯甲烷:甲醇=50:1的混合液洗涤,所得滤液经减压浓缩后得到式13所示化合物((3-((吡啶-2-基甲基)氨基甲酰基)苯并[4,5]咪唑并[1,2-a]吡嗪-1-基)氨基甲酸叔丁酯),并直接用于下一步反应。
[0073]
(10)目标化合物14a(ldh-e-4)的制备:
[0074][0075]
根据上述反应式,将式13所示化合物(3-((吡啶-2-基甲基)氨基甲酰基)苯并[4,5]咪唑并[1,2-a]吡嗪-1-基)氨基甲酸叔丁酯)溶于2ml二氯甲烷dcm中,冷却至0℃后缓慢
加入三氟乙酸tfa0.2 ml,室温搅拌4小时,通过tlc显示反应完成后加入饱和碳酸氢钠水溶液至不再产生气泡,并使用二氯甲烷萃取(3
×
50ml),合并有机相,有机相经饱和食盐水洗涤(50ml)、无水硫酸钠干燥、减压浓缩、过柱,得到目标化合物ldh-e-4(1-氨基-n-(吡啶-2-基甲基)苯并[4,5]咪唑并[1,2-a]吡嗪-3-甲酰胺,45mg,70%)。产物的图谱信息如下:
[0076]1h nmr(500mhz,dmso-d6)δ8.95(s,1h),8.81(t,j=6.0hz,1h),8.56(d,j=4.8hz,1h),8.45(d,j=8.2hz,1h),7.92(d,j=8.2hz,1h),7.78(t,j=7.7hz,1h),7.56
–
7.46(m,4h),7.39(d,j=7.8hz,1h),7.30(t,j=6.2hz,1h),4.68(d,j=5.8hz,2h).
13
c nmr(126mhz,dmso-d6)δ164.2,158.2,149.4,143.6,137.3,136.3,130.6,130.2,126.4,123.6,122.8,121.9,120.7,113.6,111.5,44.6。
[0077]
实施例2ldh-e-4特异性靶向a
2a
r的特性研究
[0078]
(1)细胞培养
[0079]
本试验中所用的a2a-hek293、a1-hek293(hek293细胞购自atcc,过表达细胞株的构建具体参考“borodovsky,a.,et al.,small molecule azd4635 inhibitor ofa2ar signaling rescues immune cell function including cd103 dendritic cells enhancing anti-tumor immunity.journal for immunotherapy ofcancer,2020.8(2):p.e000417.”)、mba-md-231(购自atcc)细胞培养于含有10%胎牛血清与1%双抗(青霉素与链霉素)的dmem培养基中,4t1、pbmc细胞培养于含有10%胎牛血清与1%双抗(青霉素与链霉素)的rpim-1640培养基中,上述细胞均置于37℃,5%co2的细胞培养箱中进行培养。
[0080]
(2)化合物对a
2a
r和a1r的亲和力测试
[0081]
1)neca的配制
[0082]
①
neca母液的配制:称取0.00157gneca,溶于250μl无菌水中,分装成30μl/管,置于-20℃保存。
[0083]
②
neca工作液的配制:取10μlneca母液,加990μl ddh2o中,制成1ml。
[0084]
2)[3h]-zm 241385工作液和[3h]-dpcpx工作液配置
[0085]
①
[3h]-zm 241385和[3h]-dpcpx(购自中国同福股份有限公司)母液:取原液分装,每支5μl(原液浓度为20μm),置于-20℃保存。
[0086]
②
[3h]-zm 241385和[3h]-dpcpx工作液:3nm,[3h]-zm 241385和[3h]-dpcpx终浓度:1nm。
[0087]
3)细胞的收集、裂解、膜蛋白提取
[0088]
①
将a
2a-hek293细胞传代到10
×
10cm培养皿中,置于37℃,5%co2条件下将密度培养至90%后用于实验。取出培养皿,弃去培养液,用3ml pbs冲洗两次。
[0089]
②
往培养皿中加1mlpbs,放置3min。
[0090]
③
冲洗下细胞,然后转移至1.5ml离心管中,3500rpm、4℃离心8min。
[0091]
④
倒去上清液,加入1ml lysis buffer和pmsf(lysis:pmsf=100:1),4℃下孵育30min。
[0092]
⑤
冰浴下过针头(1ml针头)15次。
[0093]
⑥
在离心管中加至三分之二的反应液lysis buffer(约3ml),高速离心(15000rpm,4℃)20min。
[0094]
⑦
倒去上清液,加入反应液reactionbuffer 1ml,冰浴下过针头(1ml针头)15次。
在离心管中加至三分之二体积的反应液reaction buffer(约3ml),高速离心(15000rpm,4℃)20min。
[0095]
⑧
倒去上清液,将提取得到的蛋白溶于500μl反应缓冲液reactionbuffer,过针头约10次即为膜蛋白溶液。使用bca试剂测定蛋白浓度,分装后贮存于-80℃冰箱中,用于放射性配体结合实验。
[0096]
4)放射性配体结合实验
[0097]
①
加样体系如表1所示:
[0098]
表1放射性配体结合实验的加样体系
[0099][0100]
②
每管加入膜蛋白溶液(60μg/管,含ada 10μg/ml),放射性配基1nm[3h]-zm241385、1nm[3h]-dpcpx,非特异管加入终浓度为10uμ的neca,化合物(实施例1)管分别加入10-4
,10-5
,10-6
,10-7
,10-8
,10-9
,10-10
m7个不同浓度的化合物,震荡混匀,37℃下孵育30min,水浴中终止反应,使用gf/c玻璃纤维滤纸负压抽滤以分离游离配体和结合配体,用预冷的50mm tris-hcl冲洗3次,每次约1ml。将膜取下,倒扣在托盘上,烘烤3min直至烤干。将烘干的小圆片滤膜按顺序放入闪烁管中,加入2ml闪烁液。在液体闪烁计数仪中进行[3h]计数。每一结合点均做3复管,取平均值。
[0101]
5)数据处理及统计学方法
[0102]
化合物抑制率(i%)=(总结合管cpm-化合物cpm)/(总结合管cpm-非特异结合管cpm)
×
100%。采用graphpad prism软件对实验结果进行统计分析,制作图表。且ki=ic
50
/(1 (l/kd)),所有数据以均数
±
标准误表示,组间比较用one-way anova检验,取p《0.05,具有统计学意义。其中,ki是竞争配体的亲和常数,ic
50
是取代放射性配体结合50%的化合物浓度,[l]是放射性配体的自由浓度,kd是放射性配体的离解常数。
[0103]
(3)实验结果
[0104]
表2化合物与a
2a
r、a1r的亲和力
[0105][0106]
测试化合物对a
2a
r的ki可以测试其对a
2a
r的结合能力,再通过测试化合物与a1受体的ki可以测试其选择性。如表2和图1(a)的实验结果所示,该化合物对a
2a
r的ki为155.4
±
1.1pm,对a
2a
r有极高的靶向性,对a1r的ki为10.58
±
0.7nm,且对a
2a
r的亲和力是对a1r的68倍,有极高的选择性。
[0107]
实施例3ldh-e-4抑制camp累积的特性研究
[0108]
(1)化合物及缓冲液配置
[0109]
制备含有1
×
hbss(sigma)、0.1%bsa(perkin elmer)、20mm hepes(gibco)和100nm ibmx(sigma)的测定缓冲液。使用simulationbuffer配置8
×
测试化合物储备溶液(10-4
,10-5
,10-6
,10-7
,10-8
,10-9
,10-10
m)和8
×
cgs21680储备溶液(50nm)。使用lysis&detection buffer裂解缓冲液制备20
×
camp-d2和20
×
抗camp-eu3 检测试剂溶液。
[0110]
(2)htrf法测试胞内camp含量
[0111]
将hek293-a
2a
细胞接种在384孔板中,其中每孔含有38,000个细胞以及15μl测定缓冲液中。将2.5μl测试化合物溶液加入上述384孔板的指定孔中,并在37℃下温育10分钟。然后在384孔板中加入2.5μl cgs21680原液,37℃再孵育30分钟(反应体系终体积为20μl)。最后,在孔板的每孔中加入10μl camp-d2和10μl抗camp-eu3 检测试剂,室温孵育1h。在酶标仪上收集665nm和620nm波长处的数据。
[0112]
(3)实验结果
[0113]
表3化合物抑制camp累积的ic
50
[0114][0115]
腺苷与a
2a
r结合可以引起胞内camp累积,进而引起下游一系列通路激活,最终导致免疫抑制。如表3和图1(b)的实验结果所示,该化合物抑制cgs21680(50nm)引起的camp累积的ic
50
为97.2
±
4.4nm,说明其有较好的抑制camp累积的效果。
[0116]
实施例4ldh-e-4促进pbmc分泌细胞因子
[0117]
(1)pbmc分离
[0118]
1)将20ml人外周血(来自中山大学药学院的自愿者,血液由中山大学东校区校医院代抽)倒入40ml无菌pbs缓冲液中进行稀释;
[0119]
2)取50ml离心管,加入10ml ficoll溶液,并用滴管在其上层小心加入稀释好的外周血20ml;
[0120]
3)将离心管置于水平转离心机中,4℃下300g密度梯度离心20min;
[0121]
4)离心后管内分为四层,上层为血浆和pbs,最下层为红细胞,中间漂浮的絮状层为目的单个核细胞(pbmc)。
[0122]
5)使用滴管吸取中间漂浮的絮状单个核细胞,置于新的50ml离心管中,加入3倍体积的rpmi-1640,1500rpm离心10min,弃去上清获得pbmc细胞沉淀。
[0123]
(2)化合物促进pbmc释放il-2
[0124]
1)将分离的pbmc细胞接种到96孔板中,每孔20,000个细胞,并在37℃和5%co2下孵育过夜;
[0125]
2)往孔板的1640培养基中加入ldh-e-4(10-4
,10-5
,10-6
,10-7
,10-8
,10-9
,10-10
m)预孵育1h;
[0126]
3)1h后加入neca(1μm)孵育1h;
[0127]
4)1h后加入cd3、cd28(400ng/ml)激活24h;
[0128]
5)24h后收集上清,用elisa测试培养基中的il-2含量。
[0129]
(4)实验结果
[0130]
表4化合物促进il-2释放的ec
50
[0131][0132]
腺苷与a
2a
r结合可以引起免疫抑制,导致免疫细胞分泌细胞因子减少,削弱免疫细胞功能。如表4和图1(c)的实验结果所示,该化合物拮抗neca(1μm)引起的il-2释放减少的ec
50
为342.8
±
3.6nm,说明其有较好的促进免疫细胞分泌细胞因子的效果。
[0133]
实施例5ldh-e-4在共培养中对免疫细胞增加肿瘤杀伤作用的研究
[0134]
(1)pbmc的分离和激活同实施例4;
[0135]
(2)pbmc与乳腺癌细胞共培养
[0136]
1)将mda-mb-231乳腺癌细胞铺至96孔板中(20,000/孔),过夜培养;
[0137]
2)向预先铺设有mda-mb-231的孔中加入过夜激活的pbmc(100,000/孔);
[0138]
3)往1640培养基中加入ldh-e-4(1μm)预孵育1h;
[0139]
4)1h后加入neca(1μm)共培养育24h;
[0140]
5)检测培养基中的乳酸脱氢酶(ldh)含量,由细胞凋亡或坏死而造成的细胞膜结构破坏会导致细胞浆内的ldh释放到培养基中,检测培养基中ldh的相对量,即可代表pbmc导致mda-mb-231细胞的相对死亡量。
[0141]
(3)实验结果
[0142]a2a
r激动剂neca可抑制pbmc对mda-mb-231乳腺癌细胞的杀伤作用,如图2(a)的实验结果所示,ldh-e-4可恢复pbmc对mda-mb-231乳腺癌细胞的杀伤能力。
[0143]
实施例6ldh-e-4在体内模型对肿瘤生长的抑制作用研究
[0144]
(1)ldh-e-4对小鼠皮下乳腺癌细胞移植瘤模型的影响
[0145]
1)取对数生长期的乳腺癌细胞(mda-mb-231乳腺癌细胞),消化后计数,将预冷pbs与matrigel按1:1的比例混合,重悬细胞,得到浓度为3
×
105个/100μl的细胞悬液,置于冰上。在4-5周龄balb/c小鼠腹背两侧的皮下部位各注射100μl细胞悬液;
[0146]
2)待皮下肿瘤体积约为50mm3时,随机分为4组。组别及给药设置如下:对照组,每天安慰剂(即溶药所用溶剂:15%蓖麻油 85%灭菌pbs);ldh-e-4(ip)组,每天给药,腹腔注射,剂量为15mg/kg;ldh-e-4(po)组,每天给药,灌胃,剂量为15mg/kg;
[0147]
3)连续给药26天内,每天测量小鼠体重和肿瘤大小,绘制小鼠体重生长曲线;
[0148]
4)给药26天后,处死小鼠进行解剖,剥离皮下肿瘤,称重。
[0149]
(2)实验结果
[0150]
实验结果如图2(b-d)所示,图(2b)为在小鼠皮下乳腺癌细胞移植瘤模型中给药期间的肿瘤体积变化图,ldh-e-4口服(po)和腹腔注射组(ip)均能够抑制正常免疫小鼠的肿瘤生长;ldh-e-4腹腔注射组较ldh-e-4口服组能够更显著地抑制正常免疫小鼠的肿瘤生长,表明ldh-e-4腹腔注射具有更好的效果;图(2c)显示ldh-e-4口服和腹腔注射均能够显著减轻乳腺癌肿瘤的重量,其中ldh-e-4口服组的肿瘤抑制率约为43%,ldh-e-4腹腔注射组的肿瘤抑制率约为66%;图(2d)为给药26天后,剥离的乳腺癌皮下肿瘤外观图,说明ldh-e-4口服(po)和腹腔注射组(ip)均能够抑制正常免疫小鼠的肿瘤生长,其中,腹腔注射组
(ip)的抑制效果更好。
[0151]
以上对本发明的实施方式作了详细说明,但本发明不限于所描述的实施方式。对于本领域的技术人员而言,在不脱离本发明原理和精神的情况下,对这些实施方式进行多种变化、修改、替换和变型,仍落入本发明的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
