用于治疗心力衰竭的具有作为serca2a的纯或显著纯刺激剂活性的雄甾烷衍生物
技术领域
1.本发明涉及制药学领域,具体来说涉及用于治疗急性心力衰竭的雄甾烷衍生物。
背景技术:
2.心力衰竭(hf)的患病率是年龄依赖性的,范围从60岁以下人群的低于2%到75岁以上人群的超过10%(metra m&teerlink jr,lancet 2017,390:1981-1995)。大多数hf患者具有高血压、冠状动脉疾病、心肌病、瓣膜疾病或这些障碍的组合病史(metra m&teerlink jr,lancet 2017,390:1981-1995)。预计发生hf的计算终生风险会提高,并且高血压患者的风险更高(lloyd-jones dm等,circulation 2002,106:3068
–
3072)。hf患者预后不良,住院率和死亡率高。
3.hf的临床症状由心脏的双重病理特点引起,所述特点包括变力性异常,其导致心缩期排血减少(收缩功能障碍),以及顺应性异常,其中心室从静脉系统吸取血液的能力受损(舒张功能障碍)。这反过来导致可用于收缩期收缩的血液量减少(左心室(lv)充盈受损)。所述受损的收缩和松弛是由作为细胞内ca
2
储库的肌浆网(sr)的ca
2
摄入减少引起的细胞内ca
2
分布异常的结果(bers dm等,ann n.y.acad sci 2006,1080:165-177)。所述ca
2
摄入由sr膜的ca
2
atp酶(serca2a)操作,这是一种主动膜转运。serca2a活性在生理上受到它与受磷蛋白(pln)的相互作用的限制(bers dm.,annu rev physiol 2008,70:23-49;maclennan dh&kranias eg,nat rev mol cell biol 2003,4(7):566-577);这种限制通常通过pln被蛋白激酶a(pka)的磷酸化来缓解,这是一个作为hf重塑的结果而被严重抑制的信号传导途径(lohse m等,circ res 2003,93:896-906)。因此,serca2a功能在衰竭的心肌中受损(bers dm等,ann n.y.acad sci 2006,1080:165-177),并因此是sr的ca
2
摄入减少的主要原因。除了对肌细胞的收缩和松弛的影响之外,ca
2
的异常分布还会促进心律失常(zaza和rocchetti,curr pharm des 2015,21:1053-1061),并且从长远来看,它通过凋亡来加速肌细胞丧失(nakayama h等,j clin invest 2007,117:2431-44)。serca2a功能下降还提高收缩的能量成本,因为它需要通过na-ca交换器(ncx)补偿性地增加ca
2
的排出量,这个过程能量效率较低(lipskaya l等,expert opin biol ther 2010,10:29-41)。大量证据表明,serca2a功能的正常化可恢复细胞内ca
2
稳态,并在原位改善心肌细胞和心脏的收缩和松弛(byrne mj等,gene therapy 2008,15:1550-1557;sato等,jbc 2001,276:9392-99)。概括来说,在hf中serca2a功能的恢复可以改善心脏松弛并可能改善收缩,同时使心律失常、心肌耗氧量和肌细胞死亡降至最低(lipskaya l等,expert opin biol ther.2010,10:29-41)。这突出了对“纯”serca2a激活剂的需求。事实上,由于ca
2
的螯合改善,serca2a激活可以提高sr内用于产生ca
2
波的阈值,对维持所述波的ca
2
诱导的ca
2
释放施加负反馈(fernandez-tenorio m和niggli e j,mol cell cardiol 2018,119:87-95)。因此,纯或显著纯的serca2a激活可能降低心律失常的风险,并因此证明了具有serca2a刺激作用的化合物的重要性。
4.总之,能够单独增强serca2a功能的新型分子可能在hf中改善整体心功能。这为寻找具有这种药效学特征的新化合物提供了强烈动机。
5.当前的hf长期疗法旨在预防“心肌重塑”(例如β-阻断剂、ace抑制剂和醛固酮拮抗剂),所述心肌重塑是对收缩性下降的慢性适应不良响应,会扩大初始损伤并成为疾病演变的基础(heineke j和molkentin d,nat rev 2006,7:589-600)。尽管这种方法具有无可争议的优点,但它并未针对作为定义hf并造成其症状的功能错乱的受损的“收缩”和“松弛”。事实上,特别是在疾病晚期,提高心肌收缩/松弛的药物(“变力性/松弛性药剂”)仍被广泛使用,并且对患者管理至关重要(metra m和teerlink jr,lancet 2017,390:1981-1995)。这些药物包括拟交感神经胺类(多巴酚丁胺)和作为具有强血管舒张作用的ca
2
敏化剂的左西孟旦。不幸的是,这些药剂通过具有潜在有害成分的机制起作用,例如促进威胁生命的心律失常、提高心肌耗氧量以及由于血管舒张引起的血压下降而造成的已经不足的冠状动脉血流的受损(ashkar h,makaryus an statpearls.treasure island(fl):statpearls publishing,2018年1月-2017年12月19日(https://www.ncbi.nlm.nih.gov/books/nbk470431/);gong b.等,j cardiothorac vasc anesth 2015,29:1415-25editorial)。这将变力性药剂的使用限制到疾病晚期,从而失去了在疾病过程早期提高收缩的潜在益处。此外,这些药剂不能改善患者的预后和存活率,并且它们的治疗性使用必须被仔细监测(ashkar h&makaryus an,statpearls.treasure island(fl):statpearls publishing,2018年1月-2017年12月19日)(gong b.等,j cardiothorac vasc anesth 2015,29:1415-25editorial)。
6.在正性变力性药物中,强心苷地高辛这种na
/k
atp酶的酶活性的抑制剂,是过去最常处方的药物之一。然而,在过去几十年中它的使用越来越少,这是因为难以将地高辛维持在地高辛在其中表现出有益效果的血清浓度范围(0.5-0.7ng/ml)之内而不达到0.9ng/ml的阈值水平,在高于所述阈值水平时已观察到主要由心律失常造成的死亡风险提高(packer m,journal of cardiac failure 2016,22:726-730;packer m,eur j heart failure 2018,20:851-852)。
7.对于具有正性变力性以外的作用机制的hf药物的开发,也正在进行深入的研究。在许多药物中,研究最多且正在临床开发的药物是:serelaxin-重组松弛素2介导物;ularitide-重组利尿钠肽;omecamtiv mecarbil-心肌肌球蛋白激活剂;bms986231-no供体;adrecizumab-肾上腺髓质素抑制剂;anx-042-np的剪接变体;td1439-脑啡肽酶(nep)抑制剂。然而,当在2-3期临床试验中评估时,这些新药剂均未达到主要终点而没有安全性问题。
8.慢性hf(chf)患者的临床病程和预后在急性hf(ahf)发作后要差得多(solomon sd等,circulation 2007,116:1482-87)。ahfs可以被定义为hf的症状和体征的新发作或复发,需要紧急评估和治疗,并导致计划外的护理或住院治疗。一半的ahfs患者的收缩功能降低(hfref),代表了潜在的未来疗法的靶标(braunwald e.lancet 2015;385:812-24)。在射血分数降低(ref)的患者中用于ahfs的疗法聚焦于使用血管扩张剂、利尿剂或超滤来缓解充血或使用正性变力性药物提高心输出量。尽管这种治疗策略降低了心脏性猝死的风险,但在因ahfs住院的患者中出院后事件发生率仍然高得令人无法接受。可用的疗法可以导致许多不想要的心血管副作用,例如由变力性疗法引起的心肌缺血、心脏损伤和心律失常,特
别是在冠状动脉疾病(cad)患者中(abraham wt等,j am coll cardiol 2005,46:57
–
64;flaherty jd等,j am coll cardiol.2009,53(3):254-63),低血压,以及由血管扩张剂引起的外周器官(肾脏)低灌注,特别是在具有低血压的hf患者中。因此,住院期间的主要目标是在不引起心脏和/或肾脏损伤的情况下提高心输出量。此外,几乎没有关注检查或治疗受损的左心室(lv)舒张松弛,这在具有hf但保持ef的剩余的50%患者中,是导致hf症状的原因。此外,ef降低的ahfs患者也具有心室舒张受损,其对心功能的整体衰竭有贡献。已经开发出各种不同的超声心动图指标,以在hf动物模型和hf患者两者中测量心脏松弛能力(例如,早期二尖瓣环形组织速度[e']降低和早期二尖瓣流入[e]减速时间[dt]降低),以及lv充盈压提高的超声心动图参数(例如e/e'比率)。尽管在某些动物模型和患者中单个指数变化的对应关系不能完全重叠,但它们在心室松弛受损的动物模型中的整体变化确实可以转化到人类病症,并用于研究药物在ahfs中的效果(shah sa等,am heart j 2009,157:1035-41)。
[0009]
最近已经研究了提高serca2a功能的各种治疗方法。这些方法包括通过基因转移引起的serca2a过表达(byrne等,gene therapy 2008,15:1550-1557)、通过表达具有负显性的突变体来使pln失活(hoshijima m等,nat.med.2002,8:864
–
871;iwanaga y等,j clin investig 2004,113:727
–
736)、adv-shrna(suckau l等,circulation 2009,119:1241
–
1252)、microrna(等,plos one 2014,9:e92188)或抗体(kaye dm等,j.am.coll.cardiol.2007,50:253
–
260)。正如由在hf中应用serca2a基因递送的最大的iib期临床试验(cupid 2)的阴性结果所强调的,这些方法的远未解决的主要问题在于构建物递送(病毒载体等)和剂量调整(hulot js,eur heart j 2016,19:1534-1541)。最近已描述了结构上不同于istaroxime的抑制pln的小分子(吡啶酮衍生物)(kaneko m.等,eur j pharmacol 2017,814:1-7)。
[0010]
因此,小分子serca2a激活剂的开发对于治疗hf将是有利的,并且仍然代表着非常有前途的策略。
[0011]
istaroxime是一种正在临床开发中的用于治疗ahfs的新型小分子药物。istaroxime被公开在ep0825197和s.de munari等(j.med.chem.2003,64:3644-3654)中,并且是化合物(3z,5α)-3-[(2-氨基乙氧基)亚氨基]雄甾烷-6,17-二酮。istaroxime具有抑制na
/k
泵(micheletti等,jpharmacol exp ther 2002,303:592-600)并同时激活serca2a(rocchetti m等,j pharmacol exp ther.2005,313:207-15)的双重作用机制。在相同的变力性水平下,istaroxime的致心律失常作用明显低于纯na
/k
泵抑制剂地高辛(rocchetti m等,j pharmacol exp ther.2005,313:207-15)。这表明通过提高ca
2
从胞浆的清除率(alemanni,j mol cell cardiol 2011,50:910-8),serca2a刺激也可以使na
/k
泵阻断的致心律失常作用降至最低(rocchetti m等,j pharmacol exp ther.2005,313:207-15;zaza和rocchetti,curr parm des 2015,21:1053-1061),同时保持其变力性作用。在临床研究中已经证实了istaroxime降低致心律失常作用(gheorghiade m等,j am coll cardiol 2008,51:2276-85)。
[0012]
在hf患者中,istaroxime输注改善了收缩和舒张功能两者(horizon研究)(gheorghiade m等,j am coll cardiol 2008,51:2276-85;shah sa等,am heart j 2009,157:1035-41)。收缩功能的改善被检测为收缩组织速度(s')和收缩末期弹性的斜率(espvr
斜率)的增加;舒张期顺应性的提高通过舒张期组织速度(e')的增加和舒张末期弹性(edpvr斜率)的降低来揭示(shah sa等,am heart j 2009,157:1035-41)。
[0013]
尽管具有出色的药效学特征,但由于胃肠道(gi)吸收差和清除率高,istaroxime并不是长期给药的最佳选择。因此,仅将istaroxime开发用于住院ahfs患者中的静脉输注,并且其给药需要训练有素的医务人员(dec gw,j am coll cardiol.2008,51:2286-88;shah sa等,am heart j 2009,157:1035-41)。
[0014]
因此,对用于治疗hf的具有正性松弛作用并且可以优选通过口服途径给药的化合物,存在着长期需求(butler j等,eur j heart failure 2018,20:839-841;wagner s等,circ res 2015,116:1956-1970;hasenfuss g和teerlink jr.,eur heart j.2011,32(15):1838-45)。
[0015]
改进的舒张功能可能可以通过“纯”serca2a激活剂来实现。然而,尽管对发现旨在选择性激活serca2a的小分子或基因疗法进行了深入研究,但迄今为止尚未获得有希望的临床结果。
[0016]
本发明满足了上述需求并克服了现有技术的问题。
技术实现要素:
[0017]
现在已发现,某些雄甾烷衍生物表现出纯或显著纯的serca2a激活。换句话说,本文中提供的雄甾烷衍生物显著激活serca2a,但不抑制或仅仅适度地抑制na /k atp酶泵。总的来说,这些雄甾烷衍生物含有通过碳连接物附连在碳-3(c3)处的官能团和c6和/或c7处的官能团。这些纯或显著纯serca2a激活剂的结构具有本文所示的通式(i):
[0018][0019]
其中x是羧酸、羧酸酯及其生物电子等排体(硫酸酯、磺酸、磷酸酯、膦酸酯或含氮杂环例如三唑和四唑)、伯醇、醚或胺基(例如伯胺、仲胺或环胺)中的任一者;
[0020]
n是1、2、3、4或5;
[0021]
c3-c1’虚线表示c3-c1’位置处任选的环外双键c=c;
[0022]
c2-c3虚线表示任选的环内双键c=c;
[0023]
c6处的y是α-或β-构型的羟基(oh)或α-构型的羟甲基(ch2oh);
[0024]
c7处的z是-h或α-构型的-oh或酮;所述虚线表示在该位置中任选的羰基(c=o)。
[0025]
本文中公开的化合物可以包括对映异构和/或非对映异构混合物、它们的可药用盐、溶剂化物或水合物或它们的代谢物和代谢前体。
[0026]
在本发明的情形中,代谢物和代谢前体意味着已通过代谢反应被转化,但基本上维持或提高了药理活性的式(i)的化合物。
[0027]
代谢物或代谢前体的实例是羟化,羧化,磺化,糖基化,甲基化或去甲基化,乙酰化,共价连接到葡萄糖醛酸、甘氨酸和其他氨基酸、谷胱甘肽,氧化或还原的式(i)的化合物的衍生物。
[0028]
某些式(i)的化合物、特别是酯类,也可以是活性形式的前体药物。
[0029]
在式(i)的化合物可以表现出互变异构现象的情况下,所述结构式打算覆盖所有的互变异构体;本发明在其范围内包括所有可能的立体异构体、z和e异构体、光学异构体、对映异构体及其混合物。
[0030]
可药用盐也包括在本发明的范围内。可药用盐是保留基础化合物的生物活性,并源自于已知的可药用酸例如盐酸、氢溴酸、硫酸、磷酸、硝酸、延胡索酸、琥珀酸、草酸、苹果酸、酒石酸、马来酸、柠檬酸、甲磺酸或苯甲酸和本领域中常用的其他酸的盐,参见例如《制药用盐和共晶体》(pharmaceutical salts and co-crystals),主编:johan wouters,luc qu
éré
,rsc publishing,2011。
[0031]
本发明的另一个目的是所述通式(i)的化合物,其用作药物,特别是用于治疗hf。
[0032]
在某些实施方式中,本发明请求保护的化合物选自:(e)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸;(z)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸;(e)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸;(z)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸;(e)-3-[2-(氮杂环丁烷-3-基)亚乙基]-6α-羟基雄甾烷-17-酮;(z)-3-[2-(氮杂环丁烷-3-基)亚乙基]-6α-羟基雄甾烷-17-酮;(e)-3-(4-氨基丁基)-6α-羟基雄甾-2-烯-17-酮氢碘酸盐;3-[2-(哌啶-4-基)乙基]-6α-羟基雄甾-2-烯-17-酮氢碘酸盐;(ez)-3-(4-氨基亚丁基]-6α-羟基雄甾烷-17-酮;(e)-3-[2-(哌啶-4-基)亚乙基]-6α-羟基雄甾烷-17-酮;(z)-3-[2-(哌啶-4-基)亚乙基]-6α-羟基雄甾烷-17-酮;3β-[2-(哌啶-4-基)乙基]-6α-羟基雄甾烷-17-酮;(6α-羟基-17-酮基雄甾烷-3β-基)乙酸乙酯;4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸;4-(6β-羟基-17-酮基雄甾烷-3-基)丁酸;2-(6β-羟基-17-酮基雄甾烷-3-基)乙酸;4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸乙酯;4-(6α-羟基-17-酮基雄甾烷-3-基)己酸乙酯;4-(6β-羟基-17-酮基雄甾烷-3-基)己酸;(ez)-3-(5-n-甲基氨基亚戊基]-6α-羟甲基雄甾烷-7,17-二酮;(ez)-3-[2-(吡咯烷-3-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮;(ez)-3-[2-(氮杂环丁烷-2-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮;(ez)-3-[2-(哌啶-4-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮;(ez)-3-(5-n-甲基氨基亚戊基)-6α-羟甲基-7α-羟基雄甾烷-17-酮;3β-[2-(氮杂环丁烷-2-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮;3β-[2-(氮杂环丁烷-2-基)乙基]-6α-羟甲基-7α-羟基雄甾烷-17-酮;3β-[2-(吡咯烷-3-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮;3β-[2-(吡咯烷-3-基)乙基]6α-羟甲基-7α-羟基雄甾烷-17-酮;3β-[2-(哌啶-4-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮;和3β-[2-(哌啶-4-基)乙基]-6α-羟甲基-7α-羟基雄甾烷-17-酮。
[0033]
本发明的另一个目的是包含任选地与其他治疗活性成分相组合的一种或多种式(i)的化合物的药物组合物。进而,这些药物组合物可以被配制成用于口服给药、静脉内或肌肉内注射、吸入、玻璃体内注射等。在特定实施方式中,本文公开的药物组合物被用于治疗hf。
[0034]
本发明的上述和其他目的现在也将利用实施例和附图进行详细公开。
附图说明
[0035]
图1示出了在从stz大鼠分离的大鼠心室肌细胞中1μm cvie216对肌浆网(sr)ca
2
摄入参数的影响(载入方案)。所述sr ca
2
摄入参数包括ca
2
瞬时(cat)幅度(图a)、ca
2
诱导的ca
2
释放(cicr)增益(图b)和ca
2
衰减的时间常数(τ)(图c)。在图a-c中,对照(n=16-18)和cvie216(n=20-23)中的曲线之间的差异是统计显著的(p《0.05,双向anova)。
[0036]
图2示出了在从stz大鼠分离的大鼠心室肌细胞中1μm cvie214对肌浆网(sr)ca
2
摄入参数的影响(载入方案)。所述sr ca
2
摄入参数包括ca
2
瞬时(cat)幅度(图a)、ca
2
诱导的ca
2
释放(cicr)增益(图b)和ca
2
衰减的时间常数(τ)(图c)。对照(n=14)和cvie214(n=11)中的曲线之间的差异在图b中是统计显著的(p《0.05),并且在图c中接近显著(p=0.05)。
[0037]
图3示出了在豚鼠心室肌细胞中,在各种不同的刺激率(hz)下cvie216对动作电位(ap)和动作电位持续时间(apd)的短期可变性(stv)的影响。在图a中从左至右示出了在基础条件下(ctr,实心圆圈;n》13)或在1μm cvie216存在下(空心圆圈;n》11),90%复极化时的动作电位持续时间(apd
90
)(左图)、舒张期膜电位(e
diast
)(中图)和最大去极化速度(dv/dt
max
)(右图)的刺激率依赖性。对于所有参数来说,在对照组与cvie216组之间测量到的差异不是统计显著的。在图b中,示出了对照(ctr,实心圆圈)和cvie216(空心圆圈)组的stv与平均apd
90
之间的线性相关性。在每个组中将来自于所有起搏频率的数据合并,以将stv评估扩展到宽的apd范围。实线是数据点的线性拟合(对照斜率=0.013,相比于cvie216斜率=0.009,ns),以表明cvie216不改变stv对apd延长的敏感性。
[0038]
图4示出了在豚鼠心室肌细胞中,在各种不同的刺激率(hz)下cvie214对动作电位(ap)和动作电位持续时间(apd)的短期可变性(stv)的影响。在图a中从左至右示出了在基础条件下(ctr,实心圆圈;n》17)或在1μm cvie214存在下(空心圆圈;n》17),90%复极化时的动作电位持续时间(apd
90
)(左图)、舒张期膜电位(e
diast
)(中图)和最大去极化速度(dv/dt
max
)(右图)的刺激率依赖性。对于所有参数来说,在对照组与cvie214组之间测量到的差异不是统计显著的。在图b中,示出了对照(ctr,实心圆圈)和cvie214(空心圆圈)组的stv与平均apd
90
之间的线性相关性。在每个组中将来自于所有起搏频率的数据合并,以将stv评估扩展到宽的apd范围。实线是数据点的线性拟合(对照斜率=0.012,相比于cvie214斜率=0.014,ns),表明cvie214不改变stv对apd延长的敏感性。
具体实施方案
[0039]
本文公开了可用于治疗心力衰竭的组合物和方法。具体来说,本文提供了包含新型雄甾烷衍生物的组合物。此外,存在一组本文中描述的新型雄甾烷衍生物,它们激活serca2a,同时仅仅适度抑制na
/k
atp酶泵。这组被称为“显著纯”serca2a刺激剂的雄甾烷衍生物具有通式(i),并且在c3碳处带有含有胺的官能团。本文中描述的另一组新型雄甾烷衍生物表现出强烈的serca2a激活并且没有任何显著的na
/k
atp酶泵抑制。这组被称为“纯”serca2a刺激剂的雄甾烷衍生物具有通式(i),并且在c3碳处带有通过间隔物连接的含有羧酸/酯的官能团。在其他实施方式中,所述通式(i)的显著纯或纯serca2a刺激剂可以在c3碳处包括含有醇、硫酸酯或磷酸酯的官能团。因此,这些组合物具有正性松弛性特征,并且可用于选择性激活serca2a并在同时避免na
/k
atp酶泵抑制的致心律失常作用。现在将
在下文中更详细描述本文公开的组合物和方法。
[0040]
定义
[0041]
除非另有定义,否则本文中使用的所有技术和科学术语都具有与本发明所属领域的普通技术人员通常理解的相同的含义。除非另有说明,否则使用标准技术。尽管与本文所描述的类似或等同的方法和材料可以用于本公开的实践或测试,但在下文中描述了适合的方法和材料。所述材料、方法和实施例仅仅是说明性的,而不打算是限制性的。本文提到的所有出版物、专利和其他文件整体通过引用并入本文。
[0042]
当在本文中使用时,单数形式包括复数指称物,除非上下文明确指出不是如此。
[0043]
术语“约”是指由于用于获得度量的设备的典型误差率而造成的测量例如体积、时间、压力、浓度等的数值的变动。在一个实施方式中,术语“约”意味着在所报告的数值的5%以内,优选地,术语“约”意味着在所报告的数值的3%以内。
[0044]
术语“心力衰竭”是指以典型症状(例如呼吸困难、踝关节肿胀和疲劳)为特征,并可能伴有由结构性和功能性心脏异常引起的体征(例如颈静脉压升高、肺裂音和外周性水肿),导致在休息时或紧张期间心输出量降低和/或心内压升高的临床综合征。
[0045]
术语“急性心力衰竭”或“ahf”在本文中可互换使用,并且通常是指hf的症状和/或体征的快速发作或恶化,需要立即治疗和住院。当前对“急性心力衰竭”的定义是相当非特异性的,并可能包括具有以不同的临床表现、病因、诱发因素、治疗方法和预后为特征的几种表型的广谱病症。另外,大部分患者具有亚急性病程,并可能在入院前几天发生hf的体征和症状的逐渐恶化。
[0046]
术语“慢性心力衰竭”或“chf”在本文中可互换使用,并且是指基于hf的体征和症状的存在以及左心室射血分数(lvef)进行的慢性hf的当前临床分类,其分为三类:“伴有射血分数降低的心力衰竭”或“hfref”,其特征是lvef小于约40%;“伴有中程射血分数的心力衰竭”或“hfmef”或“hfmref”,其特征是lvef为约40%至约49%;以及“保留射血分数的心力衰竭”或“hfpef”,其特征是lvef等于或大于约50%。术语“hfmref”和“hfpef”包括两个附加标准;即利尿钠肽水平提高(bnp》35pg/ml和/或nt-probnp》125pg/ml)并伴有结构性和/或功能性心脏病的证据(左心室肥大和/或左心房扩大和/或舒张功能障碍的证据)。仅在“hfref”患者中证实了hf的基于证据的药物的疗效,而在“hfpef”中没有任何治疗表现出结果的显著改善。
[0047]
术语“代谢物”和“代谢前体”是指已经通过代谢反应被转化/修饰,但基本上维持或表现出药理活性增加的化合物。
[0048]
术语“治疗”是指疾病或病症的治疗或改善的任何成功标志。治疗可以包括例如降低或减轻所述疾病或病症的一种或多种症状的严重性,或者可以包括降低个体例如人类患者所经历的疾病、缺陷、障碍或不良状况等的症状的频率。
[0049]
术语“预防”是指在个体例如人类患者中阻止疾病或病症例如急性心力衰竭。例如,如果处于发生心力衰竭的风险中的个体用本发明的方法进行治疗并且在晚些时候未发生心力衰竭,则已在该个体中预防了所述疾病。
[0050]
术语“治疗或预防”有时在本文中用于指称引起疾病或病症的某种水平的治疗或改善的方法,并且考虑到了针对该目的的一系列结果,包括但不限于所述病症的完全预防。
[0051]
当在本文中使用时,术语“可药用载体”意味着活性化合物例如具有通式(i)的雄
甾烷衍生物或其代谢物可以与它们合并,并且在合并后可用于将所述化合物给药到哺乳动物的化学组合物。
[0052]
当在本文中使用时,术语“可药用”盐、溶剂化物、水合物或酯意味着与药物组合物的任何其他成分相容,对所述组合物待给药的对象无害的活性成分的盐、溶剂化物、水合物或酯形式。术语“可药用盐”也指保留了基础化合物的生物活性并且源自于可药用酸的化合物的盐形式。
[0053]
当在本文中用于指称测量心功能时,术语“参数”意味着可以使用本领域中可用的适合测量技术观察或测量的任何心功能。心功能的示例性“参数”的非限制性名单包括钙瞬时幅度(cat),钙诱导的钙释放(cicr),钙衰减的时间常数,90%复极化时的动作电位持续时间(apd
90
)、舒张期膜电位(e
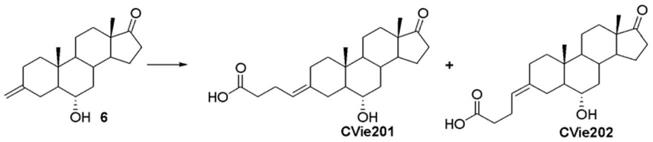
diast
)、最大去极化速度(dv/dt
max
),心率,血压,舒张期松驰,收缩期收缩,左心室射血分数(lvef),舒张压,收缩压,心输出量,每搏输出量,收缩速度(s’),早期松弛速度(e’),晚期松弛速度(a’),左心室充盈压指数(e/e’),e波减速时间(dt),二尖瓣减速指数(dt/e),减速斜率(e/dt),心脏指数,二尖瓣流入速度等。正如本领域普通技术人员会认识到的,测量心功能的一个或多个“参数”可用于与平均正常“参数”相比检测心脏功能障碍,并且也可用于确定在治疗后或治疗期间心功能是否改善。
[0054]
术语“显著纯”在涉及serca2a激活或刺激时,是指具有在无细胞体系(来自于豚鼠、狗、大鼠等的sr心脏微粒体)中以统计显著的方式刺激serca2a活性的能力,同时在无细胞体系中仅轻微抑制纯化的狗肾na /k atp酶(即具有大于约0.5μm、优选地大于约1μm的ic
50
)的化合物,例如雄甾烷衍生物。
[0055]
术语“纯”在涉及serca2a激活或刺激时,是指具有在无细胞体系(来自于豚鼠、狗、大鼠等的sr心脏微粒体)中以统计显著的方式刺激serca2a活性的能力,并且不表现出na /k atp酶泵的显著抑制(即具有大于约100μm的ic
50
)的化合物,例如雄甾烷衍生物。
[0056]
术语“治疗活性”或“活性”成分或化合物是指为所述物质给药到的个体提供有益效果的物质。“治疗有效量”或“治疗有效剂量”是足以为所述组合物或活性成分给药到的个体提供有益效果的组合物或活性成分的量。
[0057]
具有显著纯或纯serca2a刺激活性的雄甾烷衍生物
[0058]
本发明是基于具有显著纯或纯serca2a刺激活性的雄甾烷衍生物的发现。换句话说,它们是表现出serca2a刺激和仅仅轻微或无na
/k
atp酶泵抑制的衍生物。
[0059]
这些新的雄甾烷衍生物在c-3碳处用带有各种不同的官能团例如含有胺或含有羧酸/酯的官能团的碳连接物官能化。此外,这些新的雄甾烷衍生物也在c-6和/或c-7碳处用例如羟基、羟甲基、或酮基官能化。优选地,适合用于本发明的每种新型雄甾烷衍生物具有通式(i):
[0060][0061]
其中x是羧酸、羧酸酯和它们的生物电子等排体(硫酸酯、磺酸、磷酸酯、膦酸酯或
含氮杂环例如三唑和四唑)、伯醇、醚或胺基(例如伯胺、仲胺或环胺)中的任一者;
[0062]
c6处的碳连接物具有用n表示的一个或多个碳,n是1至5之间的整数(例如1、2、3、4或5);
[0063]
虚线表示任选的双键(c3-c1’或c2-c3处的c=c)和c7处的c=o;
[0064]
c6处的y基团是α-或β-构型的羟基(oh)或α-构型的羟甲基(ch2oh);并且
[0065]
c7处的z基团可以是-h或α-构型的-oh或酮(c=o)。
[0066]
在某些实施方式中,可能需要显著纯serca2a刺激剂。就此而言,适合用于此的雄甾烷衍生物可以包括其中x是胺官能团(例如伯胺、仲胺或环胺)的具有通式(i)的衍生物。然而,在某些实施方式中,可能希望选择纯serca2a刺激剂。就此而言,适合使用的雄甾烷衍生物可以包括那些具有通式(i)的衍生物,只是x不是胺官能团(例如伯胺、仲胺或环胺)。在优选实施方式中,所述纯serca2a刺激剂具有通式(i)并且在x处带有羧酸或羧酸酯。
[0067]
优选地,本文公开的雄甾烷衍生物在z或y或两者处含有含氧官能团。
[0068]
也适合用于本发明的是由通式(i)表示的化合物的对映异构和/或非对映异构混合物,以及它们的可药用盐、溶剂化物和/或水合物和它们的代谢物和/或代谢前体。代谢物或代谢前体的实例包括羟化,羧化,磺化,乙酰化,糖基化,葡萄糖醛酸化,甲基化或去甲基化,共价连接到谷胱甘肽、甘氨酸或其他氨基酸,氧化或还原的式(i)化合物的衍生物。此外,某些式(i)的化合物、特别是酯类,也可以是活性形式的前体药物。可药用盐的实例包括但不限于盐酸、氢溴酸、硫酸、磷酸、硝酸、延胡索酸、琥珀酸、草酸、苹果酸、酒石酸、马来酸、柠檬酸、甲磺酸或苯甲酸和本领域中常用的其他酸(参见例如《制药用盐和共晶体》(pharmaceutical salts and co-crystals),主编:johan wouters,luc qu
éré
,rsc publishing,2011,其全部内容通过引用并入本文)。
[0069]
在式(i)的化合物可以表现出互变异构现象的情况下,所述结构式打算覆盖所有的互变异构体,包括但不限于所有可能的立体异构体、z和e异构体、光学异构体、对映异构体及其混合物。
[0070]
适合用于本发明的具体雄甾烷衍生物包括:
[0071]
(e)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸(cvie201)
[0072][0073]
(z)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸(cvie202)
[0074][0075]
(e)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸(cvie203)
[0076]
[0077]
(z)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸(cvie204)
[0078][0079]
(e)-3-[2-(氮杂环丁烷-3-基)亚乙基]-6α-羟基雄甾烷-17-酮(cvie205)
[0080][0081]
(z)-3-[2-(氮杂环丁烷-3-基)亚乙基]-6α-羟基雄甾烷-17-酮(cvie206)
[0082][0083]
(e)-3-(4-氨基丁基)-6α-羟基雄甾-2-烯-17-酮氢碘酸盐(cvie207)
[0084][0085]
3-[2-(哌啶-4-基)乙基]-6α-羟基雄甾-2-烯-17-酮氢碘酸盐(cvie208)
[0086][0087]
(ez)-3-(4-氨基亚丁基]-6α-羟基雄甾烷-17-酮(cvie209)
[0088][0089]
(e)-3-[2-(哌啶-4-基)亚乙基]-6α-羟基雄甾烷-17-酮(cvie210)
[0090][0091]
(z)-3-[2-(哌啶-4-基)亚乙基]-6α-羟基雄甾烷-17-酮(cvie211)
[0092][0093]
3β-[2-(哌啶-4-基)乙基]-6α-羟基雄甾烷-17-酮(cvie212)
[0094][0095]
(6α-羟基-17-酮雄甾烷-3β-基)乙酸乙酯(cvie213)
[0096][0097]
4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸(cvie214)
[0098][0099]
4-(6β-羟基-17-酮基雄甾烷-3-基)丁酸(cvie215)
[0100][0101]
2-(6β-羟基-17-酮基雄甾烷-3-基)乙酸(cvie216)
[0102][0103]
4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸乙酯(cvie217)
[0104][0105]
4-(6α-羟基-17-酮基雄甾烷-3-基)己酸乙酯(cvie218)
[0106][0107]
4-(6β-羟基-17-酮基雄甾烷-3-基)己酸(cvie219)
[0108][0109]
(e,z)-3-(5-n-甲基氨基亚戊基]-6α-羟甲基雄甾烷-7,17-二酮(cvie401)
[0110][0111]
(e,z)-3-[2-(吡咯烷-3-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮(cvie402)
[0112][0113]
(e,z)-3-[2-(氮杂环丁烷-2-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮(cvie403)
[0114][0115]
(e,z)-3-[2-(哌啶-4-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮(cvie405)
[0116][0117]
(e,z)-3-(5-n-甲基氨基亚戊基)-6α-羟甲基-7α-羟基雄甾烷-17-酮(cvie406)
[0118][0119]
3β-[2-(氮杂环丁烷-2-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮(cvie407)
[0120][0121]
3β-[2-(氮杂环丁烷-2-基)乙基]-6α-羟甲基-7α-羟基雄甾烷-17-酮(cvie408)
[0122][0123]
3β-[2-(吡咯烷-3-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮(cvie409)
[0124][0125]
3β-[2-(吡咯烷-3-基)乙基]6α-羟甲基-7α-羟基雄甾烷-17-酮(cvie410)
[0126][0127]
3β-[2-(哌啶-4-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮(cvie411)
[0128][0129]
3β-[2-(哌啶-4-基)乙基]-6α-羟甲基-7α-羟基雄甾烷-17-酮(cvie412)
[0130][0131]
本发明的目的还包括利用所述式(i)的化合物的serca2a激活性质来治疗、改善、逆转或废除或减轻与serca2a激活减少相关的疾病例如心力衰竭(ahf和/或chf)的症状,或预防所述疾病。由于有缺陷的细胞内ca
2
分布在心肌重塑过程中有影响,因此通过serca2a刺激对其进行校正可能会抵消它。因此,可以防止收缩性的初始和补偿性错乱演变为明显的心力衰竭。
[0132]
正如上文提到的,本文公开的雄甾烷衍生物充当纯或显著纯serca2a激活剂。正如在下文实施例中示出的,这些化合物表现出serca2a激活。纯serca2a激活剂例如cvie201-204和cvie213-219不显著抑制na
/k
atp酶。例如,这些化合物对分离的犬肾na
/k
atp酶表现出大于100μm的ic
50
值(参见实施例3)。另一方面,显著纯serca2a激活剂例如cvie205-212和cvie401-412仅仅轻微抑制na
/k
atp酶。例如,这些化合物对分离的犬肾na
/k
atp酶表现出至少0.8μm的ic
50
值;优选地,它们对分离的犬肾na
/k
atp酶具有至少1μm的ic
50
值(实施例3)。此外,所述显著纯serca2a激活剂与istaroxime相比表现出低约6倍至约170倍的na
/k
atp酶抑制。
[0133]
在某些实施方式中,所述显著纯和纯serca2a激活剂可以通过它们附连到c3碳连接物的相应官能团(即式(i)中的x)来区分。在某些实施方式中,所述纯serca2a激活剂在c3
碳连接物处具有羧酸或羧酸酯。在其他实施方式中,所述显著纯serca2a激活剂在c3碳连接物处具有胺官能团(例如伯胺、仲胺或环胺)。
[0134]
本文中提供的纯或显著纯serca2a激活剂化合物可用于治疗心力衰竭。这种激活serca2a而不显著抑制na
/k
atp酶的能力允许这些化合物为心脏提供松弛作用以改善心功能,而不提高与na
/k
atp酶抑制相关的心律失常或心肌细胞损伤的风险。因此,这些化合物可用作治疗心力衰竭(急性或慢性)的药物,并用于治疗或预防心力衰竭的方法中。因此,可以使用完全在本领域普通技术人员能力范围之内的合成和配制技术,将它们包括在被配制以用于不同给药途径的药物组合物中。现在,将更详细地讨论利用本文中公开的纯或显著纯serca2a激活剂的药物组合物和治疗性治疗方法。
[0135]
药物组合物
[0136]
作为治疗剂的式(i)的化合物可以单独地或作为药物制剂(组合物)的组分给药。因此,本文中公开了一种药物组合物,其包含与至少一种可药用载体和/或赋形剂混合的式(i)的化合物或本文中公开的任何特定衍生物。所述药物组合物可以被配制成用于肠胃外、局部、皮下、肌肉内、口服或通过局部给药例如通过气溶胶或透皮给药到个体。在特定实施方式中,所述给药途径是口服。
[0137]
所述药物组合物可以以任何方式配制,并且取决于所述病症或疾病和患病程度、每位患者的总体医学状况、得到的优选给药方法等,可以以各种不同的单位剂型给药。用于配制和给药的技术的详情充分描述在科学和专利文献中,参见例如最新版的《remington制药学》(remington's pharmaceutical sciences,mack publishing co,easton pa)。
[0138]
所述化合物可以被配制成以用于人类或兽药的任何方便的方式给药。所述组合物中也可以存在润湿剂、乳化剂和润滑剂,例如月桂基硫酸钠和硬脂酸镁,以及着色剂、脱模剂、包衣剂、甜味剂、调味剂和增香剂、缓冲剂、防腐剂和抗氧化剂。
[0139]
根据本发明的组合物的制剂包括适合于口服、经鼻、局部、肠胃外例如通过肌肉内或静脉内注射、直肠、皮下和/或阴道内给药的制剂。所述制剂可以方便地以单位剂型存在,并且可以通过药学领域中公知的任何方法来制备。可以与载体材料组合以产生单一剂型的活性成分的量将根据待治疗的对象和/或具体给药方式而变。可以与载体材料组合以产生单一剂型的活性成分的量通常是产生治疗效果的化合物的量。
[0140]
本文提供的药物制剂可以根据本领域已知的用于药物制造的任何方法来制备。这样的制剂可以含有甜味剂、调味剂、着色剂和防腐剂。可以将制剂与适合于制造的无毒可药用赋形剂混合。制剂可以包含一种或多种稀释剂、乳化剂、防腐剂、缓冲剂、赋形剂等,并且可以以诸如液体、粉剂、乳液、冻干粉剂、喷雾剂、霜剂、洗剂、受控释放制剂、片剂、丸剂、凝胶、在贴片上、在植入物中等的形式提供。
[0141]
用于口服给药的药物制剂可以使用本领域公知的可药用载体,以合适且适合的剂量配制。这样的载体使药物能够配制成单位剂型例如片剂、凝胶片、丸剂、粉剂、糖衣丸、胶囊、液体、含片、凝胶、糖浆剂、浆液、悬液等,适合于被患者摄入。用于口服使用的药物制剂可以如下配制:使用固体赋形剂,在需要时添加适合的其他化合物后任选地研磨得到的混合物并加工所述颗粒混合物,以获得片剂或糖衣丸核心。适合的固体赋形剂是碳水化合物或蛋白质填充剂,包括例如糖类,包括乳糖、蔗糖、甘露糖醇或山梨糖醇;来自于玉米、小麦、大米、马铃薯或其他植物的淀粉;纤维素,例如甲基纤维素、羟丙基甲基纤维素或羧甲基纤
维素钠;树胶,包括阿拉伯胶和黄耆胶;以及蛋白质,例如明胶和胶原蛋白。可以添加崩解剂或增溶剂,例如交联聚乙烯吡咯烷酮、琼脂、藻酸或其盐(例如藻酸钠)。
[0142]
糖衣丸核心被提供有适合的包衣,例如浓糖溶液,其也可以含有阿拉伯胶、滑石粉、聚乙烯吡咯烷酮、卡波姆凝胶、聚乙二醇、二氧化钛、清漆溶液和/或适合的有机溶剂或溶剂混合物。可以将染料或颜料添加到所述片剂或糖衣丸包衣中,以用于产品识别或表征活性化合物的量(即剂量)。用于实践本文提供的用途和方法的药物制剂还可以口服使用,利用例如由明胶制成的推入配合型胶囊(push-fit capsule),以及由明胶和包衣例如甘油或山梨糖醇制成的软密封胶囊。推入配合型胶囊可以含有与填充剂或粘合剂例如乳糖或淀粉、润滑剂例如滑石粉或硬脂酸镁以及任选的稳定剂混合的活性药剂。在软胶囊中,可以将所述活性药剂溶解或悬浮在含有或不含稳定剂的适合液体例如脂肪油、液体石蜡或液体聚乙二醇中。
[0143]
水性悬液可以含有与适合于制造水性悬液的赋形剂混合的活性药剂(例如用于实践本文提供的用途和方法的组合物)。这样的赋形剂包括悬浮剂,例如羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、藻酸钠、聚乙烯吡咯烷酮、黄耆胶和阿拉伯胶;以及分散剂或润湿剂,例如天然存在的磷脂(例如卵磷脂)、环氧烷与脂肪酸的缩合产物(例如聚氧乙烯硬脂酸酯)、环氧乙烷与长链脂肪醇的缩合产物(例如十七亚乙基氧基鲸蜡醇)、环氧乙烷与源自于脂肪酸和己糖醇的偏酯的缩合产物(例如聚氧乙烯山梨糖醇单油酸酯)或环氧乙烷与源自于脂肪酸和己糖醇酐的偏酯的缩合产物(例如聚氧乙烯失水山梨糖醇单油酸酯)。所述水性悬液还可以含有一种或多种防腐剂例如对羟基苯甲酸乙酯或对羟基苯甲酸正丙酯、一种或多种着色剂、一种或多种调味剂和一种或多种甜味剂,例如蔗糖、阿斯巴甜或糖精或赤藓糖醇或莱鲍迪苷a。可以调整制剂的摩尔渗透压浓度。
[0144]
基于油的药物对于给药适合于本文提供的用途和方法的疏水性活性药剂来说特别有用。基于油的悬液可以通过将活性药剂悬浮在植物油例如花生油、橄榄油、芝麻油或椰子油或矿物油例如液体石蜡或它们的混合物中来配制。参见例如美国专利号5,716,928,其描述了使用精油或精油组分来提高口服给药的疏水性药物化合物的生物可利用性并降低个体间和个体内变差;也参见美国专利号5,858,401。所述油悬液可以含有增稠剂,例如蜂蜡、硬石蜡或鲸蜡醇。可以添加甜味剂以提供适口的口服制剂,例如甘油、山梨糖醇或蔗糖、赤藓糖醇或莱鲍迪甙a。这些制剂可以通过添加抗氧化剂例如抗坏血酸来保存。作为可注射油性介质的示例,参见minto j.,pharmacol.exp.ther.1997,281:93-102。本文提供的药物制剂也可以是水包油乳液的形式。所述油相可以是如上所述的植物油或矿物油或它们的混合物。适合的乳化剂包括天然存在的树胶例如阿拉伯胶和黄耆胶,天然存在的磷脂例如大豆卵磷脂,酯类,或源自于脂肪酸和己糖醇酐的偏酯例如失水山梨糖醇单油酸酯,以及这些偏酯与环氧乙烷的缩合产物例如聚氧乙烯失水山梨糖醇单油酸酯。所述乳液也可以含有甜味剂和调味剂,如在糖浆和酏剂的配制中那样。这样的制剂还可以含有缓和剂、防腐剂或着色剂。
[0145]
根据本发明,所述药物化合物还可以通过鼻内、眼内和阴道内途径给药,包括栓剂、吹入剂、粉剂和气溶胶制剂(例如甾类吸入剂,参见rohatagi,j.clin.pharmacol.1995,35:1187-1193;tjwa,ann.allergy asthma immunol.1995,75:107-111,其每一者的内容整体通过引用并入本文)。栓剂制剂可以通过将药物与适合的无刺激性赋形剂混合来制备,所
述赋形剂在常温下为固体,但在体温下为液体,因此将在体内融化以释放出所述药物。这样的材料是可可脂和聚乙二醇。
[0146]
根据本发明,所述药物化合物可以被配制成涂药棒、溶液、悬液、乳液、凝胶、霜剂、软膏、糊剂、胶冻、涂料、粉剂和气溶胶,通过局部途径透皮递送。
[0147]
根据本发明,式(i)的药物化合物可以通过吸入来递送;例如,在可选实施方式中,用于吸入的式(i)的化合物被制备成用于干式分散,例如通过将含有活性成分即式(i)的化合物的溶液喷雾干燥,例如使用在美国专利号6,509,006、6,592,904、7,097,827和6,358,530中描述的方法,每个所述专利的内容整体通过引用并入本文。示例性的干粉赋形剂包括低分子量碳水化合物或多肽,将其与式(i)的化合物混合以帮助分散。在可选实施方案中,可作为载体用于干粉分散的药物赋形剂的类型包括稳定剂例如人血清白蛋白(hsa),其也是有用的分散剂;增量剂,例如碳水化合物、氨基酸和多肽;ph调节剂或缓冲剂;盐类例如氯化钠;等等。这些载体可以是结晶或无定形形式,或者可以是两者的混合物。可用于递送粉剂或气溶胶制剂的装置包括例如在美国专利号5,605,674和7,097,827中描述的装置。
[0148]
根据本发明,所述药物化合物也可以作为纳米粒子或微球递送,用于在体内缓慢释放。例如,纳米粒子或微球可以通过在皮下缓慢释放的药物的真皮内或皮下注射来给药,参见rao j.,biomater,sci.polym.ed.1995,7:623-645;作为可生物降解和可注射的凝胶制剂来给药,参见例如gao,pharm.res.1995,12:857-863;或作为用于口服给药的微球来给药,参见例如eyles,j.pharm.pharmacol.1997,49:669-674,每个所述文献的全部内容整体通过引用并入本文。
[0149]
根据本发明,式(i)的药物化合物可以肠胃外给药,例如通过肌肉内(im)或静脉内(iv)给药,或给药到体腔或器官内腔中。这些制剂可以包含溶解在可药用载体中的活性药剂的溶液。可以使用的可接受的介质和溶剂是水、右旋糖水溶液和林格氏溶液,后者是等渗氯化钠溶液。此外,可以使用无菌不挥发油作为溶剂或悬浮介质。为此目的,可以使用任何温和的不挥发油,包括合成的甘油单酯或甘油二酯。另外,脂肪酸例如油酸同样可以用于注射剂的制备。这些溶液是无菌的,并且通常不含不想要的物质。这些制剂可以通过常规的公知灭菌技术进行灭菌。所述制剂可以含有接近生理条件所需的可药用辅助物质,例如ph调节剂和缓冲剂、毒性调节剂,例如乙酸钠、氯化钠、氯化钾、氯化钙、乳酸钠等。这些制剂中活性药剂的浓度可以在宽范围内变化,并且将根据所选的特定给药方式和患者的需要,主要基于体液量、粘度、体重等进行选择。对于iv给药来说,所述制剂可以是无菌注射制剂,例如无菌可注射水性或油性悬液。这种悬液可以使用适合的分散剂或湿润剂和悬浮剂来配制。所述无菌注射制剂也可以是在无毒的肠胃外可接受的稀释剂或溶剂例如1,3-丁二醇溶液中的悬液。给药可以通过快速浓注或连续输注(例如在特定时间段内基本上不间断地引入到血管中)来进行。
[0150]
本文提供的药物化合物和制剂可以被冷冻干燥。提供了包含本文提供的组合物的稳定的冻干制剂,其可以通过将包含本文提供的药物和填充剂例如甘露醇、海藻糖、棉子糖和蔗糖或其混合物的溶液冷冻干燥来制备。还存在许多其他常规的冻干剂。在糖中,乳糖是最常用的。还使用柠檬酸、碳酸钠、edta、苯甲醇、甘氨酸、氯化钠等(参见例如journal of excipients and food chemistry第1卷,第1期(2010)第41-54页;美国专利申请公布号20040028670)。
[0151]
治疗方法
[0152]
根据本发明,本文提供的式(i)的化合物可用于预防性和/或治疗性治疗。在治疗性应用中,将药物组合物以治疗有效量给药到已患有病症或疾病的对象。在其他实施方式中,将本文提供的药物组合物在需要的个体中以足以治疗、预防或改善所述病症或疾病的量给药。用于此用途的剂量安排和有效量(即“给药方案”)将取决于各种不同因素,包括疾病或病症的阶段、疾病或病症的严重程度、患者健康的总体状态、患者的身体状况、年龄等。在计算患者的给药方案时,还应考虑给药方式。
[0153]
在特定实施方式中,所述式(i)的化合物用于治疗患有心力衰竭的个体。在优选实施方式中,所述个体表现出急性心力衰竭的症状或已被诊断为患有急性心力衰竭。尽管所述个体可以是非人类动物,但在优选实施方式中,所述个体是人类患者,例如患有心力衰竭的人类患者。在其他实施方式中,本文提供的化合物用于在个体中刺激serca2a。
[0154]
通常,本文描述的式(i)的化合物和药物组合物可用于治疗心力衰竭或急性心力衰竭。治疗方法包括提供或呈现患有心力衰竭或急性心力衰竭的个体。在某些情况下,首先进行测量步骤以确定所述个体的基线心功能。所述测量步骤可以包括测量一种或多种心功能参数,例如但不限于心率、血压、舒张期松驰、收缩期收缩、左心室射血分数(lvef)、舒张压、收缩压、心输出量、每搏输出量、减速斜率(e/dt)、收缩速度(s’)、早期松弛速度(e’)、晚期松弛速度(a’)、左心室充盈压指数(e/e’)、e/ea或e/a比率、ea比率、e波减速时间(dt)、二尖瓣减速指数(dt/e)、减速斜率(e/dt)、心脏指数、二尖瓣流入速度等。在患有心力衰竭或心功能受损的个体中,所述测量到的参数可以包括心率降低、心脏压力降低、收缩和/或舒张压降低、左心室舒张/收缩末期容积和功能(lvef)降低或e/ea或e/a比率提高、ea比率降低、每搏输出量降低中的一者或多者。所述测量步骤也可用于确定所述药物组合物的给药的有效性(即心功能的恢复或部分恢复),和/或在治疗期间监测所述个体的状况。因此,所述测量步骤可以在所述药物组合物给药之前、期间或之后进行。正如本领域普通技术人员会认识到的,在所述测量步骤时,本领域中可用的任何适合的测量技术均适合用于本发明,并且选择对应于感兴趣的参数的适合的测量技术完全在这些专业技术人员的能力范围之内。适合的测量设备/技术的非限制性名单包括血液测试、超声心动图(包括组织多普勒成像)、心导管插入术、核压力测试、cat扫描、放射性核素心室描记扫描、听诊器、血压计等。例如,舒张期松驰可以通过超声心动图或pcwp来测量。
[0155]
本文公开的方法还包括向所述个体给药治疗有效量的通式(i)的化合物。在优选实施方式中,所述化合物在药物组合物中,例如上文讨论的任一组合。所述化合物以本文中别处公开的治疗有效剂量例如约1mg/kg至约20mg/kg之间的剂量给药。在更优选实施方式中,给药途径是口服。所述测量步骤可以在所述给药步骤之前、期间或之后进行。例如,可能希望在治疗期间和随后的一段时间内连续监测一种或多种心功能参数。
[0156]
所述给药方案也将本领域中公知的药代动力学参数考虑在内,即所述活性药剂的吸收、生物可利用性、代谢、清除的速率等(参见例如hidalgo-aragones(1996)j.steroid biochem.mol.biol.58:611-617;groning(1996)pharmazie 51:337-341;fotherby(1996)contraception 54:59-69;johnson(1995)j.pharm.sci.84:1144-1146;rohatagi(1995)pharmazie 50:610-613;brophy(1983)eur.j.clin.pharmacol.24:103-108;最新的《remington制药学》,同上)。当前技术水平允许临床医生针对每个个体患者、活性药剂和所
治疗疾病或病症确定给药方案。为用作药物的类似组合物提供的指南可作为指导,用于确定被给药以实践本文中提供的方法的给药方案、即剂量方案和剂量水平是正确且适当的。
[0157]
取决于患者所需和耐受的剂量和频率,可以进行制剂的单次或多次给药。所述制剂应当提供足够量的活性药剂,以有效治疗、预防或改善本文中所描述的病症、疾病或症状。例如,用于实践本文提供的方法和用途的组合物的用于口服给药的示例性药物制剂的每日量,可以是每日约1至约20、50、100或1000或更多μg/kg体重之间或等同量的其可药用盐、溶剂化物或水合物。
[0158]
在可选实施方式中,给药到需要的个体的式(i)的化合物的有效量或其可药用盐、溶剂化物或水合物的等同量,包括使用各种不同的给药计划,例如:a)在ahfs的情况下,为了挽救住院患者,可以将式(i)的化合物通过静脉内输注给药12h、24h、48h、72h或更长时间,并使用每分钟0,1或0,5至约10、50或100或更多μg/kg体重范围内的剂量;b)在从ahfs挽救并出院的患者中,维持治疗效果的给药计划可以是1、10、50或100或1000或更多μg/kg体重之间的每日量。
[0159]
可用于实践本发明的式(i)的化合物可以被给药,以作为单次快速浓注递送1ng/kg至50mg/kg体重之间的剂量,或递送1μg至约20mg之间的口服或静脉内剂量,或以重复方式递送,或其组合,正如专业技术人员容易确定的。在某些实施方式中,所述剂量包括以每日计或以另一种适合的周期方案计至少0.05mg/kg、0.1mg/kg、或至少0.2mg/kg、或至少0.3mg/kg、或至少0.4mg/kg、或至少0.5mg/kg、或至少0.6mg/kg、或至少0.7mg/kg、或至少0.8mg/kg、或至少0.9mg/kg、或至少1mg/kg、或至少2mg/kg、或至少3mg/kg、或至少4mg/kg、或至少5mg/kg、或至少6mg/kg、或至少7mg/kg、或至少8mg/kg、或至少9mg/kg、或至少10mg/kg、或至少15mg/kg、或至少20mg/kg、或至少25mg/kg、或至少30mg/kg、或至少35mg/kg、或至少40mg/kg、或至少45mg/kg、或至少50mg/kg。
[0160]
在一个实施方式中,本发明设想了以治疗有效剂量静脉内或皮下给药本文中描述的通式(i)的化合物,所述治疗有效剂量是约0.125mg/kg至约10mg/kg之间,例如0.125mg/kg、0.25mg/kg、0.5mg/kg、0.75mg/kg、1mg/kg、1.25mg/kg、1.5mg/kg、1.75mg/kg、2mg/kg、2.25mg/kg、2.5mg/kg、2.75mg/kg、3mg/kg、3.25mg/kg、3.5mg/kg、3.75mg/kg、4mg/kg、4.25mg/kg、4.5mg/kg、4.75mg/kg、5mg/kg、5.25mg/kg、5.5mg/kg、5.75mg/kg、6mg/kg、6.25mg/kg、6.5mg/kg、6.75mg/kg、7mg/kg、7.25mg/kg、7.5mg/kg、7.75mg/kg、8mg/kg、8.25mg/kg、8.5mg/kg、8.75mg/kg、9mg/kg、9.25mg/kg、9.5mg/kg、9.75mg/kg或10mg/kg。在优选实施方式中,所述化合物以约0.25mg/kg至约5mg/kg之间的治疗有效剂量,通过静脉内或皮下递送方式(例如注射或输注)给药。在另一个实施方式中,所述治疗有效剂量在约0.5mg/kg至约5mg/kg之间。在又一个实施方式中,所述治疗有效剂量在约0.5mg/kg至4mg/kg之间或约0.5mg/kg至约3mg/kg之间。
[0161]
在另一个实施方式中,本发明设想了以治疗有效剂量肌肉内给药本文中描述的具有通式(i)的化合物,所述治疗有效剂量在约0.25mg/kg至约50mg/kg之间,例如0.25mg/kg、0.5mg/kg、1mg/kg、1.5mg/kg、2mg/kg、2.5mg/kg、3mg/kg、3.5mg/kg、4mg/kg、4.5mg/kg、5mg/kg、5.5mg/kg、6mg/kg、6.5mg/kg、7mg/kg、7.5mg/kg、8mg/kg、8.5mg/kg、9mg/kg、9.5mg/kg、10mg/kg、10.5mg/kg、11mg/kg、11.5mg/kg、12mg/kg、12.5mg/kg、13mg/kg、13.5mg/kg、14mg/kg、14.5mg/kg、15mg/kg、15.5mg/kg、16mg/kg、16.5mg/kg、17mg/kg、17.5mg/kg、18mg/kg、
18.5mg/kg、19mg/kg、19.5mg/kg、20mg/kg、20.5mg/kg、21mg/kg、21.5mg/kg、22mg/kg、22.5mg/kg、23mg/kg、23.5mg/kg、24mg/kg、24.5mg/kg、25mg/kg、26mg/kg、27mg/kg、28mg/kg、29mg/kg、30mg/kg、31mg/kg、32mg/kg、33mg/kg、34mg/kg、35mg/kg、36mg/kg、37mg/kg、38mg/kg、39mg/kg、40mg/kg、41mg/kg、42mg/kg、43mg/kg、44mg/kg、45mg/kg、46mg/kg、47mg/kg、48mg/kg、49mg/kg或50mg/kg。在优选实施方式中,将雄甾烷衍生物以约0.25mg/kg至约35mg/kg之间的治疗有效剂量通过肌肉内递送方式(例如注射)给药。在另一个实施方式中,所述治疗有效剂量在约0.25mg/kg至30mg/kg之间。在又一个实施方式中,所述治疗有效剂量在约0.25mg/kg至10mg/kg之间。在其他实施方式中,所述治疗有效剂量在约0.25mg/kg至5mg/kg之间。
[0162]
在又一个实施方式中,本发明设想了以治疗有效剂量玻璃体内给药本文中描述的具有通式(i)的化合物,所述治疗有效剂量在约1μg至约10mg之间,例如1μg、1.25μg、1.5μg、1.75μg、2μg、2.25μg、2.5μg、2.75μg、3μg、3.25μg、3.5μg、3.75μg、4μg、4.25μg、4.5μg、4.75μg、5μg、5.25μg、5.5μg、5.75μg、6μg、6.25μg、6.5μg、6.75μg、7μg、7.25μg、7.5μg、7.75μg、8μg、8.25μg、8.5μg、8.75μg、9μg、9.25μg、9.5μg、9.75μg、10μg、20μg、30μg、40μg、50μg、60μg、70μg、80μg、90μg、100μg、150μg、200μg、250μg、300μg、350μg、400μg、450μg、500μg、550μg、600μg、650μg、700μg、750μg、800μg、850μg 900μg、950μg、1mg、1.1mg、1.2mg、1.3mg、1.4mg、1.5mg、1.6mg、1.7mg、1.8mg、1.9mg、2mg、2.1mg、2.2mg、2.3mg、2.4mg、2.5mg、2.6mg、2.7mg、2.8mg、2.9mg、3mg、3.5mg、4mg、4.5mg、5mg、5.5mg、6mg、6.5mg、7mg、7.5mg、8mg、8.5mg、9mg、9.5mg或10mg;优选地,所述剂量在约1μg至约2,000μg之间,例如约1μg至约2,000μg、或约100μg至约1,500μg、或约500μg至约1,200μg、或约500μg至约1,000μg。在某些实施方式中,通过玻璃体内给药递送的化合物的治疗有效剂量为至少约0.02mg,例如至少约0.02mg、0.03mg、0.04mg、0.05mg、0.06mg、0.07mg、0.08mg、0.09mg、0.1mg、0.15mg、0.2mg、0.25mg、0.3mg、0.35mg、0.4mg、0.45mg、0.5mg、0.55mg、0.6mg、0.65mg、0.7mg、0.75mg、0.8mg、0.85mg、0.9mg、0.95mg或1mg。
[0163]
在另一个实施方式中,本发明设想了以治疗有效剂量口服给药本文中描述的具有通式(i)的化合物,所述治疗有效剂量在约1mg/kg至约20mg/kg之间,例如1mg/kg、1.5mg/kg、2mg/kg、2.5mg/kg、3mg/kg、3.5mg/kg、4mg/kg、4.5mg/kg、5mg/kg、5.5mg/kg、6mg/kg、6.5mg/kg、7mg/kg、7.5mg/kg、8mg/kg、8.5mg/kg、9mg/kg、9.5mg/kg、10mg/kg、10.5mg/kg、11mg/kg、11.5mg/kg、12mg/kg、12.5mg/kg、13mg/kg、13.5mg/kg、14mg/kg、14.5mg/kg、15mg/kg、15.5mg/kg、16mg/kg、16.5mg/kg、17mg/kg、17.5mg/kg、18mg/kg、18.5mg/kg、19mg/kg、19.5mg/kg或20mg/kg。在优选实施方式中,所述化合物以约1mg/kg至约10mg/kg之间的治疗有效剂量通过口服递送方式给药。例如,在一个特定实施方式中,将具有通式(i)的化合物以约1和5mg/kg的剂量口服递送到人类。在某些实施方式中,本文中描述的口服剂量被一次性给药。在其他实施方式中,它被每日给药。
[0164]
在另一个实施方式中,将治疗有效量的本文中描述的具有通式(i)的化合物按照给药计划通过输注给药到个体,例如约0.1μg/kg/min至约5.0μg/kg/min,例如0.1μg/kg/min、0.2μg/kg/min、0.3μg/kg/min、0.4μg/kg/min、0.5μg/kg/min、0.6μg/kg/min、0.7μg/kg/min、0.8μg/kg/min、0.9μg/kg/min、1.0μg/kg/min、1.1μg/kg/min、1.2μg/kg/min、1.3μg/kg/min、1.4μg/kg/min、1.5μg/kg/min、1.6μg/kg/min、1.7μg/kg/min、1.8μg/kg/min、1.9
μg/kg/min、2.0μg/kg/min、2.1μg/kg/min、2.2μg/kg/min、2.3μg/kg/min、2.4μg/kg/min、2.5μg/kg/min、2.6μg/kg/min、2.7μg/kg/min、2.8μg/kg/min、2.9μg/kg/min、3.0μg/kg/min、3.1μg/kg/min、3.2μg/kg/min、3.3μg/kg/min、3.4μg/kg/min、3.5μg/kg/min、3.6μg/kg/min、3.7μg/kg/min、3.8μg/kg/min、3.9μg/kg/min、4.0μg/kg/min、4.1μg/kg/min、4.2μg/kg/min、4.3μg/kg/min、4.4μg/kg/min、4.5μg/kg/min、4.6μg/kg/min、4.7μg/kg/min、4.8μg/kg/min、4.9μg/kg/min或5.0μg/kg/min。例如,在某些实施方式中,所述化合物以有效剂量通过输注给药,所述有效剂量为约0.2μg/kg/min至约2.0μg/kg/min,或约0.2μg/kg/min至约1.5μg/kg/min,或约0.25μg/kg/min至约1.0μg/kg/min,或约0.5μg/kg/min至约1.0μg/kg/min。
[0165]
在可选实施方式中,给药到需要的个体的式(i)的化合物或其可药用盐、溶剂化物或水合物的等同物的有效量,在肺毛细血管楔压(pcwp)、组织多普勒成像(tdi)测量、呼吸困难、外周和肺静脉充血、尿液量、运动能力、血清生物标志物例如nt-probnp和高敏感性心脏肌钙蛋白(hs-ctnt)的监测的基础上个性化设置。
[0166]
在可选实施方式中,给药到需要的个体的式(i)的化合物或其可药用盐、溶剂化物或水合物的等同物的量足以维持正常的运动耐力而没有呼吸困难。
[0167]
在可选实施方式中,有效量通过pcwp、端坐呼吸、阵发性夜间呼吸困难的减少、运动耐力的提高、周围和肺静脉充血例如肺爆裂音或罗音的减少、踝关节肿胀的减少、生物标志物尿液输出量例如nt-probnp和高敏感性心肌肌钙蛋白(hs-ctnt)的减少来证实。
[0168]
在可选实施方式中,当在血流中或iv或im给药(与例如口服、通过吸入或皮下给药相反),例如作为iv或im给药或给药到体腔中或器官内腔中时,使用较低剂量的式(i)的化合物或其可药用盐、溶剂化物或水合物的等同物。在局部、喷雾、吸入或口服给药或通过粉剂、喷剂或吸入给药中,可以使用实质上更高的剂量。制备肠胃外或非肠胃外可给药的制剂的实际方法对于本领域技术人员来说是已知的或显而易见的,并在诸如remington's(参见《remington制药学》(remington's pharmaceutical sciences,mack publishing co,easton pa))的出版物中更详细描述。
[0169]
在特定实施方式中,式(i)的化合物或其可药用盐、溶剂化物或水合物的等同物被长期给药,例如从诊断之日直至患者生命的最后一天或直至疾病减轻。在可选实施方式中,从治疗期到维持期需要通过所述疾病的特定的常规已知生物标志物或临床体征的定期监测进行剂量调整。
[0170]
在可选实施方式中,在评估治疗效果、治疗方案或特定剂量或确定是否应给予治疗或维持剂量时,应该对个体例如受ahf或chf影响的患者进行日常定期筛查,以检查是否存在器官和组织牵连或损害或其程度,例如心脏(心室扩张、第三心音心脏肥大)、疲劳、疲倦、运动耐力降低、运动后恢复时间增加、肾脏(肾功能不全、少尿)、肺(端坐呼吸、阵发性夜间呼吸困难、呼吸急促)、踝关节肿胀、颈静脉压升高。在心血管疾病特别是ahf或chf的治疗中,应当以由专家选择的时间间隔进行彻底的身体检查,这些检查应集中在心脏、肺和外周循环功能上。因此,在可选实施方式中,使用式(i)的化合物或其可药用盐、溶剂化物或水合物的等同物的疗法应尽可能早、优选地在急诊室中进行,以预防症状的迅速发展,并在患者出院后持续多年,优选为患者的整个生命期间或至少与hf中使用其他药物的方式相一致的一段时间。
[0171]
根据本发明,本文提供的用途和方法可以进一步包括与其他药物或药剂共同给药。实际上,本发明选择性地校正压低的心脏生化功能(即serca2a活性)。这无疑有助于缓解现有的hf临床症状,并且与可用疗法相比具有更少的不良副作用(仅仅因为上面提到的选择性)。然而,由于chf和ahf是复杂的临床综合征,因此本发明可能与现有和未来的药物类别和/或特定药物相结合,例如:a)药物类别,例如ace抑制剂、airb、利尿剂、ca
2
通道阻断剂、β-阻断剂、毛地黄、no供体、血管扩张剂、serca2a刺激剂、脑啡肽酶(nep)抑制剂、肌球蛋白丝激活剂、重组松弛素-2介导物、重组np蛋白、可溶性鸟苷酸环化酶(sgc)的激活剂、血管紧张肽ⅱ受体的β-抑制蛋白配体;b)特定药物:氢氯噻嗪,呋塞米,维拉帕米,地尔硫卓,卡维地洛,美托洛尔,肼苯哒嗪,依普利酮,螺内酯,赖诺普利,雷米普利,硝化甘油,硝酸酯,地高辛,缬沙坦,奥美沙坦,替米沙坦,坎地沙坦,氯沙坦,诺欣妥,omecamtiv,沙库比曲,serelaxin,乌拉立肽,左西孟旦,cinaciguat。
[0172]
本发明的化合物当用作治疗剂、特别是用于治疗hf时,可以与用于治疗同一疾病的其他治疗剂组合。示例性的其他治疗剂是利尿剂例如呋塞米、布美他尼和托拉塞米,美托拉宗,醛固酮拮抗剂例如螺内酯或依普利酮,噻嗪利尿剂例如氢氯噻嗪、美托拉宗和氯噻酮。其他药剂是ace抑制剂,例如赖诺普利和雷米普利。血管紧张肽ⅱ受体阻断剂(arb)例如缬沙坦、坎地沙坦和氯沙坦,也可以考虑在内。血管紧张肽受体/脑啡肽酶抑制剂(arni)例如沙库比曲,被包括在内。其他药剂可以选自β-阻断剂例如卡维地洛和美托洛尔,或血管扩张剂例如任选地与二硝酸异山梨酯组合的肼苯哒嗪、肼苯哒嗪、硝酸酯例如硝酸甘油、氨氯地平和非洛地平、非二氢吡啶类例如地尔硫卓或维拉帕米。如果需要,本发明的化合物也可以与地高辛组合。其他药物例如伊伐布雷定和其他抗凝药,也可以考虑。另外,其他药物可能包括omecamtriv mecarbil。
[0173]
本发明的化合物可以与其他治疗剂组合,特别是可用于治疗心血管疾病、更特别是在hf的组合疗法中使用的药剂。所述组合的活性成分可以按照由医生决定的不同方案进行给药。根据本发明的实施方案,组合疗法可以通过将式(i)的化合物与一种或多种其他治疗活性成分在同时或在不同时间给药来进行。在同时给药的情况下,本发明的化合物和所述一种或多种其他活性成分可以各自配制在相应的药物组合物中。在这种情况下,本发明提供了一种特别是用于治疗心力衰竭的药剂盒,其包含分别含有本发明的化合物和所述一种或多种其他活性成分的分开的药物组合物。在另一个实施方式中,本发明提供了一种特别是用于治疗hf的单位剂型药剂盒,其包含本发明的化合物和所述一种或多种其他活性成分。
[0174]
纳米粒子、纳米脂质粒子和脂质体
[0175]
还提供了包含本文中提供的化合物的纳米粒子、纳米脂质粒子、囊泡和脂质体膜,以例如将本文中提供的药物活性化合物和组合物(式(i)的化合物或其可药用盐、溶剂化物或水合物的等同物)递送到需要的对象。在可选实施方式中,这些组合物被设计成靶向特定分子,包括生物分子例如多肽,包括细胞表面多肽,例如用于靶向所需细胞类型例如肌细胞或心脏细胞、内皮细胞等。
[0176]
提供了包含用于实践本公开的方法的化合物的多层脂质体,如在park等人的美国专利公布号20070082042中所述,其内容整体通过引用并入本文。所述多层脂质体可以使用包含角鲨烯、甾类、神经酰胺、中性脂质或油、脂肪酸和卵磷脂的油相组分混合物制备成粒
径为约200至5000nm,以捕获用于实践本文提供的用途和方法的组合物。
[0177]
脂质体可以使用如在美国专利号4,534,899、美国专利公布号20070042031中所述的任何方法来制造,包括通过包封根据本发明的活性药剂(或活性药剂的组合)来生产脂质体的方法,所述方法包括在第一储器中提供水性溶液;在第二储器中提供有机脂质溶液,然后将所述水性溶液与所述有机脂质溶液在第一混合区域中混合以产生脂质体溶液,其中所述有机脂质溶液与所述水性溶液混合以基本上瞬时产生包封所述活性药剂的脂质体;然后立即将所述脂质体溶液与缓冲溶液混合,以产生稀释的脂质体溶液。
[0178]
在一个实施方式中,用于实践本文提供的用途和方法的脂质体组合物包含取代的铵和/或聚阴离子,以例如用于将用于实践本文中提供的方法的式(i)的化合物或其可药用盐、溶剂化物或水合物的等同物靶向递送到所需细胞类型,如美国专利公布号20070110798中所述。
[0179]
提供了包含用于实践本文提供的用途和方法的根据本发明的化合物的纳米粒子,所述纳米粒子是含有活性药剂的纳米粒子的形式(例如次级纳米粒子),如在美国专利公布号20070077286中所述。在一个实施方式中,提供了包含用于实践本文提供的用途和方法的脂溶性活性药剂或脂增溶的水溶性活性药剂的纳米粒子,以与二价或三价金属盐一起起作用。
[0180]
在一个实施方式中,可以使用固体脂质悬液来配制用于实践本文提供的用途和方法的组合物,并将其递送到体内、体外或离体的哺乳动物细胞,如在美国专利公布号20050136121中所述。
[0181]
所述用于实践本文提供的用途和方法的组合物和制剂可以利用脂质体或纳米脂质体来递送。通过使用脂质体,特别是在所述脂质体的表面带有特异性针对靶细胞的配体或以其他方式偏好性地指向特定器官的情况下,人们可以聚焦于将所述活性药剂体内递送到靶细胞。参见例如美国专利号6,063,400、6,007,839;al-muhammed,j.microencapsul.1996,13:293-306;chonn,curr.opin.biotechnol.1995,6:698-708;ostro,am.j.hosp.pharm.1989,46:1576-1587。
[0182]
递送介质
[0183]
在可选实施方式中,可以使用任何递送介质来实践本文中提供的用途和方法,例如将本文中提供的化合物递送到需要的对象。例如,可以使用包含聚阳离子、阳离子型聚合物和/或阳离子型肽类例如聚乙烯亚胺的递送介质,正如在美国专利公布号20060083737中所描述的。
[0184]
在一个实施方式中,使用干燥的多肽-表面活性剂复合物来配制用于实践本文中提供的用途和方法的组合物,正如在美国专利公布号20040151766中所描述的。
[0185]
在一个实施方式中,用于实践本文中提供的用途和方法的组合物,可以使用具有细胞膜渗透肽偶联物的介质施加到细胞,正如在美国专利号7,306,783、6,589,503中所描述的。在一种情况下,将所述待递送的组合物偶联到细胞膜渗透肽。在一个实施方式中,将所述待递送的组合物和/或递送介质偶联到转运介导肽,正如在美国专利号5,846,743中所描述的,所述专利描述了高碱性并结合到聚磷酸肌醇的转运介导肽。
[0186]
在一个实施方式中,使用电通透化作为主要或辅助手段将所述组合物递送到细胞,这可以使用任何电穿孔系统,例如在美国专利号7,109,034、6,261,815、5,874,268中所
描述的。
[0187]
式(i)的化合物的制备
[0188]
本发明的化合物可以通过有机化学领域的技术人员可用的许多方法来合成。下文描述了用于制备本发明的化合物的通用和示例性合成方案。这些方案是说明性的,并且不意味着限制本领域技术人员可用于制备本文公开的化合物的可能技术。对于本领域技术人员来说,制备本发明的化合物的不同方法是显而易见的。另外,为了得到所需的一种或多种化合物,所述合成中的各种不同步骤可以以可选的顺序进行。
[0189]
按照通用方案中描述的方法制备的本发明的化合物的实例,提供在后文中阐述的实施例部分中。
[0190]
正如本领域技术人员认识到的,本发明的化合物可以使用下文描述的方法与合成有机化学领域中已知的合成方法或对其作出的变更一起来合成。反应在适合于所使用的试剂和材料并适合于待实施的转化的溶剂或溶剂混合物中进行。有机合成领域的技术人员应当理解,所述分子上存在的官能团应该与提出的转化相符。
[0191]
此外,本领域技术人员可以容易地改变在下面的方案中示例的试剂和反应条件,以包括上文中所定义的取代基的任何组合。此外,专业技术人员可以容易地为每个合成过程使用可互换的步骤,并且并入被视为必需的分离和/或纯化步骤。
[0192]
可用于制备本发明的化合物的起始材料和中间体是可商购的,或者可以通过公知的合成程序来制备。
[0193]
通过下文描述的合成获得的最终产物可以使用本领域技术人员通常已知的技术来纯化,例如制备型层析、薄层层析、hplc或结晶。
[0194]
本文中描述了用于合成本发明的化合物的示例性过程。
[0195]
在下面的制备中,除非另有陈述,否则化学化合物、溶剂、反应物和任何其他材料来自于商业来源。通常,式(i)的化合物可以从脱氢表雄酮(普拉睾酮)开始,通过多步合成来制备。脱氢表雄酮是一种商品化产品,或者可以从4-雄烯-3,17-二酮(雄烯二酮)开始按照公知的方法来制备。
[0196]
5α-雄甾烷-3β,6α,17β-三醇的制备
[0197][0198]
如de munari等(j.med.chem.,2003,46(17):3644-54)中所描述,适用于合成6-α-3,17-雄甾烷二酮(2)的中间体从脱氢表雄酮1通过硼氢化和随后的氧化来生产。简单来说,将脱氢表雄酮1(5g,17.5mmol,1当量)在thf(85ml)中的溶液在-20℃和ar下搅拌。然后向所述搅拌的溶液添加thf中的1m bh3·
thf络合物(44ml,44mmol,2.5当量),并将搅拌在室温继续3小时。小心地逐滴添加h2o(85ml),然后逐滴添加nabo3·
4h2o(5.4g,35mmol,2当量)。在室温搅拌过夜后,将所述混合物过滤。将固体用thf洗涤,然后舍弃。将清液用nacl饱和并用thf萃取(3
×
40ml)。将合并的有机萃取液在nacl和na2so4上干燥、过滤并蒸发至干。将5α-雄甾烷-3β,6α,17β-三醇2粗产物从etoac/meoh(2/1,10ml/g)结晶,得到白色固体(3.8g,70%)。
[0199]
6α-羟基雄甾烷-3,17-二酮的制备
[0200][0201]
中间体3从2通过c3和c17位置处的选择性氧化来获得。在0℃下向5α-雄甾烷-3β,6α,17β-三醇2(2g,6.5mmol,1当量)在二噁烷/h2o/吡啶(54/10/1ml)中的搅拌的溶液添加nbs(3.4g,19.5mmol,3当量)。在添加后,允许所述混合物升温至室温并搅拌过夜。将所述橙色溶液用水(50ml)稀释并用na2s2o3(350mg)淬灭。在真空下蒸发掉有机溶剂,直至白色固体出现。将所述固体过滤并用水洗涤。在40℃下干燥后,获得作为白色固体的6α-羟基雄甾烷-3,17-二酮3(1.3g,70%)。
[0202]
雄甾烷-3-亚甲基-17-酮的合成
[0203][0204]
然后通过在c3羰基上的选择性维蒂希反应和随后与5-戊烯酸的交叉复分解偶联,将6-α-3,17-雄甾烷二酮3转变成外甲烷衍生物6(雄甾烷-3-亚甲基-17-酮)。在-5℃下,向甲基三苯基溴化膦(1,66g,6mmol,4当量)在thf(10ml)中的悬液添加叔丁醇钾(670mg,6mmol,4当量)。所述溶液立即变色成亮橙色。10分钟后,添加6α-羟基雄甾烷-3,17-二酮3(450mg,1.5mmol,1当量),同时保持温度低于0℃。在添加后,立即通过添加1m hcl水溶液(15ml)将反应淬灭,并用etoac萃取(3x20ml)。将合并的有机相在na2so4上干燥并蒸发至干。将所述粗萃取物通过柱层析进行纯化(洗脱剂etoac:石油精4:6),产生376mg(83%)作为白色泡沫的雄甾烷-3-亚甲基-17-酮6。
[0205]
通过交叉复分解反应从前体6直接合成cvie 201和202
[0206][0207]
向雄甾烷-3-亚甲基-17-酮6(100mg,0.33mmol,1当量)在dcm(1ml)中的溶液添加hoveyda-grubbs第二代催化剂(12mg,0.015mmol,0.05当量)。然后将所述溶液在回流下加热,并且每20分钟用10μl 4-戊烯酸处理(总共330μl,3.3mmol,10当量)。在添加结束后,将混合物继续回流2h。将反应混合物真空浓缩并通过快速层析进行纯化(洗脱剂丙酮:石油精3:7 0.1%hco2h),以获得两种不同的白色固体(e)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸(4.8mg,4%)(cvie201)和(z)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸(7.2mg,6%)(cvie202)。
[0208]
或者,通过修改维蒂希反应来获得cvie201和cvie202。在一种方法(路线a)中,利用极性溶剂例如dmso和碱例如nah使甜菜碱中间体稳定。第二种方法(路线b)允许使用非质
子性溶剂例如thf作为碱使氧杂磷杂环丁烷中间体稳定。路线a产生非对映异构体的混合物(60%的z/顺式cvie202;30%的e/反式cvie201),而路线b提供源自于氧杂磷杂环丁烷中间体的cvie202。任一程序都需要产生如下所述的非对映异构体7和/或8。
[0209]
cvie 201和202通过维蒂希反应的可选合成路线a
[0210][0211]
在ar气氛下向无水dmso(1ml)小心地添加矿物油中的60%nah(100mg,2.56mmol,8当量)。将得到的溶液在60℃搅拌20分钟。在室温冷却后,添加(3-羧基丙基)三苯基溴化膦(550mg,1.28mmol,4当量)。立即出现亮橙色颜色。将溶液搅拌2h。然后向所述混合物添加6α-羟基雄甾烷-3,17-二酮3(100mg,0.32mmol,1当量)。允许得到的溶液在室温继续搅拌4h。将反应混合物用etoac(25ml)稀释,并用1m hcl水溶液(3x 30ml)洗涤。将有机层在na2so4上干燥并蒸发至干,得到25mg粗制品。
[0212]
将所述粗制品首先溶解在meoh(1.5ml)中,然后添加edc盐酸盐(115mg,0.6mmol,2当量)和dmap(5mg,0.03mmol,0.1当量)。将所述溶液在室温搅拌3h。在真空浓缩后,将粗品固体溶解在etoac(15ml)中并用1m hcl水溶液(3x 10ml)洗涤。将粗产物在硅胶上通过快速层析进行纯化(丙酮:石油精3:7),得到25mg包含非对映异构体7和8的混合物的透明油状物(20%)。
[0213]
路线b
[0214][0215]
在ar气氛和-40℃下,向(3-羧基丙基)三苯基溴化膦(8.5g,20mmol,6当量)的无水thf(33ml)悬液小心地添加thf中的1m lihmds溶液(40ml,40mmol,12当量)。将所述溶液在-40℃搅拌过夜,直至出现亮橙色。然后在-40℃下向所述溶液添加6α-羟基雄甾烷-3,17-二酮3(1g,3.3mmol,1当量)。在室温搅拌过夜后,将反应混合物用1m hcl水溶液(300ml)淬灭并用etoac(3x 350ml)萃取。将合并的有机层在na2so4上干燥并蒸发至干。
[0216]
将所述粗制品溶解在无水etoh(17ml)中,然后添加edc盐酸盐(1.26mg,6.6mmol,2当量)和dmap(50mg,0.3mmol,0.1当量)。允许所述混合物在室温搅拌3h。将反应在etoac(150ml)中稀释并用1m hcl水溶液(3x 100ml)洗涤。将粗产物在硅胶上通过快速层析进行纯化(丙酮:石油精3:7),得到910mg(72%)化合物8。
[0217][0218]
甲基(或乙基)酯的最终水解
[0219]
向甲基酯7和8(25mg,0.06mmol,1当量)在thf(600μl)和水(200μl)中的溶液添加
1m lioh水性溶液(150μl,2.5当量)。2h后,将反应用水(10ml)稀释,并通过添加1m hcl直至溶液达到ph 1进行淬灭。将水性相用etoac(3x 15ml)萃取。将合并的有机层在na2so4上干燥并蒸发至干。将粗品通过快速层析进行纯化(acoet:石油精7:3 1%hcooh)。获得两种白色固体,对应于e(7mg,31%)和z(12mg,54%)非对映异构体(分别为cvie201和cvie202)。
[0220][0221]
cvie 203和204的合成方式:中间体化合物12的合成
[0222]
为了制备cvie203和cvie204,首先从6-α-3,17-雄甾烷二酮3生产前体12。通过与乙二醇和酸催化剂(p-tsa或樟脑磺酸)的组合在甲苯中反应,将6-α-3,17-雄甾烷二酮3的羰基作为二缩酮进行保护,得到化合物9。将化合物9用pcc或其他氧化剂氧化,得到化合物10,然后将其用nabh4或kbh4还原,产生c6-羟基选择性地为β-构型的被保护的醇11。如de munari等(j.med.chem.,2003,46(17):3644-54)中所述在丙酮中通过酸性处理对所述环状二缩酮进行最后切割,得到前体12。
[0223]
简单来说,将6α-羟基雄甾烷-3,17-二酮(1.5g,4.9mmol,1当量)、乙二醇(10.5ml,88mmol,36当量)和ptsa(561mg,2.9mmol,0.6当量)在甲苯(160ml)中的溶液使用dean-stark分水器在回流下搅拌12h。在冷却至室温后,将所述混合物用5%nahco3水溶液中和。分离有机层并用h2o(2
×
40ml)洗涤,在na2so4上干燥并蒸发至干,产生作为白色固体化合物的3,3:17,17-双(亚乙基二氧)雄甾烷-6α-醇9(1.9g,98%)。
[0224]
在0℃下,向3,3:17,17-双(亚乙基二氧)雄甾烷-6α-醇(3g,14mmol,1当量)9和抗坏血酸钠(1.2g,14mmol,4当量)在无水ch2cl2(87ml)中的溶液添加pcc(148mg,0.69mmol,4当量)。将所述混合物在室温搅拌过夜。将混合物用1m hcl水溶液(3x 30ml)和水(3x30ml)洗涤。将有机层在na2so4上干燥并蒸发至干。将粗品在硅胶柱上通过快速层析进行纯化(洗脱剂丙酮:石油精2:8)。获得作为白色固体的3,3:17,17-双(亚乙基二氧)雄甾烷-6-酮10(1.53g(96%))。
[0225]
在0℃下向3,3:17,17-双(亚乙基二氧)雄甾烷-6-酮10(1g,2.5mmol,1当量)在meoh(13ml)中的搅拌的悬液添加nabh4(144mg,3mmol,1.2当量)。在0℃下2h后,逐滴添加h2o(40ml)。将所述混合物用etoac(3
×
40ml)萃取。将合并的有机萃取液在na2so4上干燥、过滤并蒸发至干,得到白色固体,其是3,3:17,17-双(亚乙基二氧)雄甾烷-6β-醇11(915mg,92%)。
[0226]
向3,3:17,17-双(亚乙基二氧)雄甾烷-6β-醇11(910mg,2.3mmol,1当量)在丙酮(46ml)中的溶液在5分钟内添加分成小份的ptsa(2.26g,11.5mmol,5当量)。在室温搅拌1h后,通过添加5%nahco3水溶液直至ph 7,将所述溶液淬灭。在搅拌5分钟后出现白色固体。在真空中除去挥发物。将悬液用ch2cl2(3x 30ml)萃取,并将合并的有机萃取液用盐水(40ml)洗涤,在na2so4上干燥,过滤并蒸发。将得到的固体与正己烷/etoac 8/2(10ml)搅拌
45分钟,然后通过过滤进行收集。将所述固体在45℃干燥3小时。得到568mg(81%)白色固体(即6β-羟基雄甾烷-3,17-二酮12)。
[0227]
12转变成最终的cvie 203和204
[0228][0229]
然后使用上文为cvie201和cvie202描述的相同程序,通过维蒂希反应从前体12获得cvie203和cvie204。利用noesy实验来鉴定两种异构体中c3-c1’双键处的构型。
[0230]
简单来说,在ar气氛下向无水dmso(1ml)小心地添加矿物油中的60%nah(100mg,2.56mmol,8当量)。将得到的溶液在60℃搅拌20分钟。在室温冷却后,添加(3-羧基丙基)三苯基溴化膦(550mg,1.28mmol,4当量)。立即出现亮橙色颜色。将所述溶液搅拌2h。然后向所述混合物添加6β-羟基雄甾烷-3,17-二酮12(100mg,0.32mmol,1当量)。允许得到的溶液在室温继续搅拌4h。将反应混合物用etoac(25ml)稀释并用1m hcl水溶液(3x 30ml)洗涤。将有机层在na2so4上干燥并蒸发至干,得到25mg粗制品。
[0231]
然后将所述粗制品溶解在meoh(1.5ml)中。添加edc盐酸盐(115mg,0.6mmol,2当量)和dmap(5mg,0.03mmol,0.1当量)。将所述溶液在室温搅拌3h。在真空浓缩后,将粗品固体溶解在etoac(15ml)中并用1m hcl水溶液(3x 10ml)洗涤。将所述粗产物在硅胶上通过快速层析进行纯化(丙酮:石油精3:7),得到非对映异构体13和14的混合物,得率分别为17%和30%。
[0232]
将反应混合物真空浓缩并通过快速层析进行纯化(洗脱剂丙酮:石油精3:7 0.1%hco2h),得到两种不同的白色固体(e)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸(cvie203)和(z)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸(cvie204)。
[0233]
通过加氢和酯水解生产cvie214、cvie215和cvie217
[0234][0235]
化合物cvie217从上述非对映异构体7 8的混合物生产。简单来说,所述非对映异构体的c3-c1’双键的加氢在etoac中使用pd-c催化来进行。得到的化合物是cvie217。c3处形成的立体中心的构型通过noesy实验来鉴定。然后将化合物cvie217用thf中的1m lioh或naoh水解,以产生cvie214。同样地,将非对映异构体13 14在etoac中使用pd-c催化进行加氢,以产生酯化合物19,然后将其用thf中的1m lioh或naoh水解,以产生cvie215。
[0236]
通过维蒂希反应和随后的c=c加氢和酯水解生产cvie213和cvie216
[0237][0238]
化合物6-α-3,17-雄甾烷二酮3也被用作通过horner-emmons反应合成cvie213和cvie216的起始点。首先,在ar气氛和0℃下,向矿物油中的60%nah(1.3g,33mmol,5当量)在dmf(200ml)中的悬液小心地添加磷酰基乙酸三乙酯(6.5ml,33mmol,5当量)。将得到的溶液在室温升温并搅拌20分钟。然后在0℃下添加6-α-3,17-雄甾烷二酮3(2g,6.5mmol,1当量)。在室温搅拌过夜后,通过小心地添加h2o(100ml)将反应淬灭,并用et2o(3x 150ml)萃取。将合并的有机层在na2so4上干燥并真空蒸发。将粗品在硅胶柱上通过快速层析进行纯化(丙酮:石油精3:7),产生2.1g(86%)两种非对映异构体的透明油状混合物(化合物21)。
[0239]
在ar气氛下,向非对映异构体化合物21(2g,5.3mmol,1当量)在etoac(200ml)中的脱气的溶液添加10%pd-c(700mg)。在三个真空/氢气循环后,允许反应在h2气氛下在室温搅拌过夜。在通过真空/ar循环除去氢气后,将反应混合物在上过滤。将过滤的溶液蒸发至干。不需纯化获得1.8g(90%)cvie213产物。用thf中的1m lioh或naoh进一步水解cvie213产生cvie216。
[0240]
通过维蒂希反应和随后的c=c加氢和酯水解生产cvie218和cvie219
[0241][0242]
同样地,将6-α-3,17-雄甾烷二酮3与适合的三苯基鏻盐(例如5-羧基三苯基溴化膦、lihmds、thf,然后是etoh(或meoh))反应,产生化合物24。接下来,在氢气存在下使用pd-c催化将化合物24催化加氢产生cvie218,其在c-3位置处包括c6链。用thf中的1m lioh或naoh将cvie218水解产生cvie219。
[0243]
通过使用boc保护的胺的复分解反应和随后的boc去保护从前体6合成具有伯胺基的衍生物
[0244][0245]
对于具有伯胺基作为式(i)中的x取代基的衍生物的合成来说,使用上文为cvie201和cvie202的合成所述的相同的实验条件对前体6进行交叉复分解反应。
[0246]
简单来说,向雄甾烷-3-亚甲基-17-酮6在dcm中的溶液添加hoveyda-grubbs第二代催化剂。然后使用适合的boc保护的胺(例如叔丁基戊-4-烯-1-基氨基甲酸酯或n-boc-4-戊炔-1-胺)将雄甾烷-3-亚甲基-17-酮6与外亚甲基合并,产生非对映异构体25(25%得
率)。将化合物25(50mg,0,1mmol,1当量)用500μl tfa/dcm(dcm中的三氟乙酸)的1:1混合物处理,然后在室温搅拌,以直接切割boc基团。在室温搅拌1分钟后,将反应用etoac(50ml)稀释并用饱和nahco3水溶液(3x30ml)洗涤。将有机相在na2so4上干燥,过滤并蒸发至干,产生作为白色固体的(ez)-3-(4-氨基亚丁基]-6α-羟基雄甾烷-17-酮(cvie209)(28mg,75%)。
[0247]
可选地,将化合物25与三甲基甲硅烷基碘化物在醇性溶剂(例如meoh)中反应,导致boc切割并伴有环外双键的迁移,以产生在c2与c3之间具有环内双键的cvie207。简单来说,在室温下向非对映异构体25(50mg,0,1mmol,1当量)的溶液添加dcm中的1m tmsi(100μl,0,1mmol,1当量)。在相同温度下搅拌2h后,在真空中除去溶剂。向残留物添加甲醇(2ml)并在室温放置1h。在真空中除去溶剂后,不需纯化即获得cvie207。
[0248]
具有环外不饱和性的环胺衍生物的合成:cvie205、cvie206、cvie210和cvie211
[0249][0250]
环胺衍生物通过如上为cvie203和cvie204所述的氢化钠(nah)-dmso维蒂希反应来合成,并在同时利用适合的n-保护的鏻盐例如n-boc-4-(2-三苯基鏻乙基)氮杂环丁烷碘化物以产生化合物26和27,或利用n-boc-3-(2-三苯基鏻乙基)哌啶碘化物以产生化合物28和29。在纯化所述非对映异构体混合物后,通过用tfa酸水解切掉n-boc基团,以产生cvie205、cvie206、cvie210和cvie211。
[0251]
具有内环不饱和性的cvie208的合成(boc去保护期间的c=c双键迁移)
[0252][0253]
如上为cvie207的合成所述,用tmsi进一步处理化合物28和29产生cvie208。
[0254]
加氢和使用tfa的boc切割以产生cvie212
[0255][0256]
可选地,将化合物28和29的双键催化加氢(h2,pd-c,etoac)以合成化合物30,然后在dcm中用tfa进行boc切割产生cvie212。
[0257]
带有6α-羟甲基雄甾烷-7,17-二酮的化合物的合成从共同中间体37开始来实现。化合物37本身从4-雄烯-3,17-二酮31开始,通过将两个酮组成部分用环状缩醛保护32并同时迁移双键,用重铬酸钠氧化烯丙基位置33,形成甲硅烷基烯醇醚35,用me3al和甲醛羟甲基化(36),以及最终在酸性条件下切割缩醛来合成。所述合成在下面的段落中更详细地描述。
[0258]
化合物32的合成:(20s,7r)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-12-烯
[0259][0260]
将雄甾-4-烯-3,17-二酮31(400.0g,1.4mol)和ptsa
·
h2o(13.3g,70.0mmol)在乙二醇(8.0l)中的混合物在100℃下搅拌,直至反应透亮。约5.0l二醇在真空下蒸馏,以使沸腾温度在80-85℃左右。将所述混合物冷却到室温。将混合物调整到ph~9。然后将混合物倾倒在冰水中。将所述混合物过滤,将固体用水洗涤、收集并用丙酮研磨,得到作为黄色固体的粗品化合物32(469.0g,89%)。
[0261]
化合物33的合成:(20s,7r)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-12-烯
[0262]-14-酮
[0263][0264]
将化合物32(440.0g,1.2mol)、hosu(541.2g,4.7mol)和na2cr2o7·
h2o(527.5g,1.8mol)在丙酮(8.0l)中的混合物在50℃剧烈搅拌2天。在冷却到室温后,将混合物用na2so3水溶液淬灭并搅拌20min。将混合物倾倒在冰水中。将得到的混合物搅拌20min,然后过滤。将固体滤出物用水洗涤、收集并在真空中干燥,得到作为黄色固体的粗品化合物33(390.0g,85%)。
[0265]
化合物34的合成:(7s,20s)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-14-酮
[0266][0267]
将化合物33(50.0g,128.9mmol)在etoac(1250ml)中的混合物添加到pd/c(16.0g)。然后将所述混合物在h2下在室温搅拌过夜。tlc显示反应完成。将混合物过滤,浓缩,并通过快速层析进行纯化(pe/ea=2/1),得到作为白色固体的化合物34(25.0g,50.0%)。
[0268]
化合物35的合成:1-((20s,7r)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-13-烯-14-基氧基)-1,1-二甲基-1-硅乙烷
[0269][0270]
将化合物34(20.0g,51.3mmol)在无水thf(100.0ml)中的混合物在-78℃下搅拌,然后逐滴添加甲苯中的1.5m lda(205.2ml,307.8mmol)。在相同温度下搅拌1小时后,逐滴添加me3sicl(50.0ml,400.1mmol)。在-70℃搅拌3小时后,将温度升高到-30℃并添加三乙胺(33.5g,331.5mmol)。在相同温度下搅拌1小时后,将混合物升温至室温并添加水(200.0ml)和etoac(100.0ml)。将分离的水性层用etoac萃取。将合并的有机层用盐水洗涤,在na2so4上干燥,过滤并蒸发至干。将残留物通过快速层析进行纯化(pe/ea=2/1),得到作为白色固体的化合物35(14.3g,60.3%)。
[0271]
化合物36的合成:(13s,20s,7r)-13-(羟甲基)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-14-酮
[0272][0273]
将2,6-二苯基酚(10.0g,27.6mmol)在无水dcm(450.0ml)中的混合物逐滴添加到me3al在甲苯中的溶液(41.4ml,82.9mmol),同时用冰/水浴冷却,使得温度不超过室温。在室温搅拌1小时后,将所述溶液在0℃冷却,并逐滴添加三氧杂环己烷(24.8g,276.0mmol)在无水dcm(100.0ml)中的溶液。将所述浅黄色溶液在0℃继续搅拌1小时,然后将温度冷却至-78℃。添加化合物35(10.0g,27.6mmol)在无水dcm(125ml)中的溶液。在-78℃搅拌1h后,将温度升高到-20℃,并将反应混合物在该温度下搅拌过夜。在室温下添加5%nahco3水溶液(85.0ml)。将所述果冻状混合物通过垫过滤,用dcm充分洗涤。将分离的有机层用水洗涤并蒸发。向残留物添加thf中的约1m tbaf(24.0ml),并将所述溶液在室温搅拌1.5h。将所述溶液用水洗涤,在na2so4上干燥,过滤并蒸发至干。将残留物通过快速层析进行纯化,得到作为黄色固体的化合物36(6.5g,71.4%)。
[0274]
化合物37的合成:(6s,10r,13s)-6-(羟甲基)-10,13-二甲基十氢-1h-环戊烷并[a]菲-3,7,17(2h,4h,8h)-三酮
[0275][0276]
将化合物36(8.0g,19.0mmol)在丙酮(100.0ml)中的混合物添加到10%hcl水溶液(50.0ml)。然后将所述混合物加热到70℃1h。tlc显示反应完成。将混合物用5%naoh水溶液淬灭,并用dcm(50.0ml
×
2)萃取。将合并的有机相用盐水(50.0ml)洗涤,在na2so4上干燥,过滤,浓缩,并通过快速层析进行纯化(dcm/ea=4/1),得到粗产物,将其用乙醚研磨,得到作为白色固体的纯产物37(3.3g,52.4%)。
[0277]
化合物38的合成:(13s,14s,20s,7r)-13-(羟甲基)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-14-醇
[0278][0279]
在0℃下向化合物36(22.0g,52.4mmol)在meoh(1000.0ml)中的混合物缓慢添加nabh4(4.0g,104.8mmol)。然后将所述混合物在室温搅拌1h。tlc显示反应完成。将混合物用5%nah2po4水溶液(220.0ml)淬灭,并用dcm(300.0ml
×
3)萃取。将合并的有机相用盐水(200.0ml)洗涤,在na2so4上干燥,过滤,浓缩,并通过快速层析进行纯化(dcm/ea=4/1),得到粗产物,将其用乙醚研磨,得到作为白色固体的化合物38(7.5g,34.1%)。
[0280]
化合物39的合成:(8s,9s,15s,2r)-9-羟基-8-(羟甲基)-2,15-二甲基四环[8.7.0.0《2,7》.0《11,15》]十七烷-5,14-二酮
[0281][0282]
向化合物38(5.7g,13.5mmol)在丙酮(70.0ml)中的混合物添加10%hcl水溶液(35.0ml)。然后,将所述混合物加热到70℃1h。tlc显示反应完成。将所述混合物用5%naoh水溶液淬灭,并用dcm(50.0ml
×
2)萃取。将合并的有机相用盐水(50ml)洗涤,在na2so4上干燥,过滤并浓缩。将残留物通过快速层析进行纯化(dcm/ea=4/1),得到粗产物,将其用乙醚研磨,得到作为白色固体的纯产物39(1.8g,40.0%)。
[0283]
化合物40的合成:(2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)氮杂环丁烷-1-甲酸叔丁酯
[0284][0285]
在-78℃下向鏻盐(2.57g,4.5mmol)在thf(25ml)中的溶液添加正丁基锂在thf中的溶液(2.5m,3.6ml,9.0mmol)。将所述混合物在30℃搅拌1小时。接下来,在-20℃下向所述混合物添加化合物37(500mg,1.5mmol),然后升温至30℃2小时。将所述混合物用饱和nh4cl(25ml)淬灭,并用etoac(25ml
×
3)萃取。将合并的有机层浓缩,并将残留物在硅胶上通过柱层析进行纯化(己烷/etoac=1/1),得到粗品化合物。将所述化合物通过反相柱进行纯化,得到作为白色固体的纯化合物40(60mg,8%)。
[0286]
化合物41的合成:3-(2-((3s,6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)氮杂环丁烷-1-甲酸叔丁酯
[0287][0288]
向化合物40(60mg,0.12mmol)在ea(3ml)中的溶液添加pd/c(60mg)。然后将所述混合物在h2下在室温搅拌过夜。将混合物过滤并将滤液浓缩,产生作为白色固体的化合物41(52mg,86%)。cvie407的合成:(3s,6s,10r,13s)-3-(2-(氮杂环丁烷-3-基)乙基)-6-(羟甲基)-10,13-二甲基十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0289][0290]
将化合物41(52mg,0.10mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌1小时。将所述混合物用饱和nahco3稀释,以调整到ph 8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,产生作为白色固体的化合物cvie407(13mg,32%)。
[0291]
化合物42的合成:3-((e)-2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)氮杂环丁烷-1-甲酸叔丁酯
[0292][0293]
在-78℃下向化合物鏻盐(514mg,0.90mmol)在thf(5ml)中的溶液添加正丁基锂在thf中的溶液(2.5m,0.7ml,1.80mmol)。将所述混合物在40℃搅拌1小时。然后,在0℃下向所
述混合物添加化合物39(100mg,0.30mmol),然后升温至40℃过夜。将反应重复9次。将混合物用饱和nh4cl(80ml)淬灭,并用etoac(100ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物42(20mg,1%)。
[0294]
cvie403的合成:(6s,10r,13s)-3-(2-(氮杂环丁烷-3-基)亚乙基)-6-(羟甲基)-10,13-二甲基十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0295][0296]
将化合物40(130mg,0.26mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌1小时。将所述混合物用饱和nahco3碱化至ph8-9。将混合物用dcm(30ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie403(13mg,得率13%)。
[0297]
化合物43的合成:3-(2-((6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-17-氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)氮杂环丁烷-1-甲酸酯
[0298][0299]
将化合物42(20mg,0.04mmol)、pd/c(10%,20mg)和pd(oh)2(20%,20mg)在etoac(2ml)中的混合物在h2下(在气球中)在室温搅拌过夜。将混合物过滤并将滤液浓缩,得到作为棕色固体的粗品化合物43(20mg,100%)。
[0300]
cvie408的合成:(6s,7s,10r,13s)-3-(2-(氮杂环丁烷-3-基)乙基)-7-羟基-6-(羟甲基)-10,13-二甲基十四氢-1h-环戊烷并[a]菲-17(2h)-酮
[0301][0302]
将化合物43(20mg,0.04mmol)在tfa/dcm(1:1,2ml)中的混合物在0℃搅拌30分钟。将所述混合物用饱和nahco3稀释,以调节到ph 8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie408(6.4mg,40%)。
[0303]
化合物44的合成:3-(2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)吡咯烷-1-甲酸叔丁酯
[0304][0305]
在-78℃下向化合物鏻盐(1.5g,2.62mmol)在thf(15ml)中的溶液添加正丁基锂在thf中的溶液(2.5m,1.57ml,3.94mmol)。将所述反应混合物在35℃搅拌1小时。然后,在-20℃下向所述混合物添加化合物37(350mg,1.05mmol)的溶液,并升温至室温2小时。将混合物用饱和nh4cl(25ml)淬灭,并用etoac(25ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过快速层析进行纯化(己烷:ea=1:1),得到粗品化合物。然后将所述化合物通过反相柱进行纯化,得到作为白色固体的纯化合物44(53mg,10%)。
[0306]
cvie402的合成:(6s,10r,13s)-6-(羟甲基)-10,13-二甲基-3-(2-(吡咯烷-3-基)亚乙基)十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0307][0308]
将化合物44(89mg,0.173mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌1小时。将所述混合物用饱和nahco3稀释,以调整到ph=8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,产生作为白色固体的化合物cvie402(38mg,53%)。
[0309]
化合物45的合成:3-(2-((3s,6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)吡咯烷-1-甲酸叔丁酯
[0310][0311]
将化合物44(53mg,0.103mmol)在ea(3ml)中的溶液添加到pd/c(60mg)。然后将所述混合物在h2下在室温搅拌过夜。将混合物过滤并将滤液浓缩,产生作为白色固体的化合物45(50mg,94%)。
[0312]
cvie409的合成:(3s,6s,10r,13s)-6-(羟甲基)-10,13-二甲基-3-(2-(吡咯烷-3-基)乙基)十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0313]
[0314]
将化合物45(50mg,0.09mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌1小时。将所述混合物用饱和nahco3稀释,以调整到ph 8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,产生作为白色固体的化合物cvie409(12mg,32%)。
[0315]
化合物46的合成:3-(2-((6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-17-氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)吡咯烷-1-甲酸叔丁酯
[0316][0317]
在-78℃下向化合物鏻盐(527mg,0.90mmol)在thf(5ml)中的溶液添加正丁基锂在thf中的溶液(2.5m,0.7ml,1.80mmol)。将所述反应混合物在35℃搅拌1小时。然后在0℃下向所述混合物添加化合物39(100mg,0.30mmol),然后升温至35℃过夜。将反应重复四次。将所述混合物用饱和nh4cl(80ml)淬灭并用etoac(100ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,得到作为白色固体的化合物46(26mg,3%)。
[0318]
化合物47的合成:3-(2-((6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-17-氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)吡咯烷-1-甲酸叔丁酯
[0319][0320]
将化合物46(26mg,0.05mmol)、pd/c(10%,30mg)和pd(oh)2(20%,30mg)在etoac(3ml)中的混合物在h2下(在气球中)在室温搅拌过夜。将混合物过滤并将滤液浓缩,得到作为黄色固体的粗品化合物47(26mg,100%)。
[0321]
cvie410的合成:(6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-3-(2-(吡咯烷-3-基)乙基)十四氢-1h-环戊烷并[a]菲-17(2h)-酮
[0322][0323]
将化合物47(26mg,0.05mmol)在tfa/dcm(1:2,2ml)中的溶液在0℃搅拌1小时。将所述混合物用饱和nahco3稀释,以调整到ph 8-9。将混合物用dcm(20ml
×
3)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie410(9mg,43%)。
[0324]
化合物48的合成:4-(2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)哌啶-1-甲酸叔丁酯
[0325][0326]
在-78℃下向化合物鏻盐(2.16g,3.60mmol)在thf(16ml)中的混合物添加正丁基锂在thf中的溶液(2.5m,2.90ml,7.20mmol)。将所述反应混合物在30℃搅拌1小时。然后在-20℃下向所述混合物添加化合物37(400mg,1.20mmol)。将混合物在-20℃搅拌30分钟,然后升温至30℃2小时。将混合物用饱和nh4cl(15ml)淬灭并用etoac(30ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,得到作为黄色固体的化合物48(28mg,4%)。
[0327]
cvie405的合成:(6s,10r,13s)-6-(羟甲基)-10,13-二甲基-3-(2-(哌啶-4-基)亚乙基)十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0328][0329]
将化合物46(80mg,0.152mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌30分钟。将所述混合物用饱和nahco3碱化至ph=8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie405(30mg,46%)。
[0330]
化合物49的合成:4-(2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)哌啶-1-甲酸叔丁酯
[0331][0332]
将化合物48(28mg,0.05mmol)和pd/c(10%,50mg)在etoac(2ml)中的混合物在h2下(在气球中)在室温搅拌过夜。将混合物过滤并将滤液浓缩,得到作为黄色固体的粗品化合物49(28mg,100%)。
[0333]
cvie411的合成:(6s,10r,13s)-6-(羟甲基)-10,13-二甲基-3-(2-(哌啶-4-基)乙基)十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0334][0335]
将化合物49(28mg,0.05mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌30分钟。将所述混合物用饱和nahco3稀释,以调整到ph 8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie411(10mg,43%)。
[0336]
化合物50的合成:4-(2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)哌啶-1-甲酸叔丁酯
[0337][0338]
在-78℃下向鏻盐(540mg,0.90mmol)在thf(5ml)中的溶液添加正丁基锂在thf中的溶液(2.5m,0.70ml,1.80mmol)。将反应混合物在40℃搅拌1小时。然后在0℃下向所述混合物添加化合物39(100mg,0.30mmol),然后升温至40℃2小时。将反应重复5次。将所述混合物用饱和nh4cl(80ml)淬灭并用etoac(100ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,得到作为白色固体的粗品化合物50(35mg,4%)。
[0339]
化合物51的合成:4-(2-((6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-17-氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)哌啶-1-甲酸叔丁酯
[0340][0341]
将化合物50(35mg,0.07mmol)、pd/c(10%,40mg)和pd(oh)2(20%,40mg)在etoac(2ml)中的混合物在h2下(在气球中)在室温搅拌过夜。将混合物过滤并将滤液浓缩,得到作为棕色固体的粗品化合物51(35mg,100%)。
[0342]
cvie412的合成:(6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-3-(2-(哌啶-4-基)乙基)十四氢-1h-环戊烷并[a]菲-17(2h)-酮
[0343]
[0344]
将化合物51(35mg,0.07mmol)在tfa/dcm(1:2,2ml)中的溶液在室温搅拌30分钟。将所述混合物用饱和nahco3稀释,以调整到ph 8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie412(13mg,46%)。
[0345]
化合物52的合成:((e)-5-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)戊基)(甲基)氨基甲酸叔丁酯
[0346][0347]
在-78℃下,向n-boc-n-甲基-5-三苯基鏻戊烯胺碘化物(4.26g,7.23mmol)在thf(50ml)中的混合物逐滴添加正丁基锂(3.18ml,7.95mmol)。将混合物在0℃搅拌20min。然后将混合物冷却至-30℃。然后向所述反应混合物添加化合物37(800mg,2.41mmol)。将所述混合物在室温搅拌过夜。将反应混合物用h2o淬灭并浓缩。将残留物在硅胶上通过柱层析进行纯化(pe/etoac=1/2),然后通过制备hplc进行纯化,产生作为无色油状物的化合物52(36mg,200mg)。
[0348]
cvie401的合成:(6s,10r,13s,e)-6-(羟甲基)-10,13-二甲基-3-(5-(甲基氨基)亚戊基)十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0349][0350]
将化合物52(60mg,0.116mmol)在tfa/dcm(1ml/2ml)中的混合物在室温搅拌过夜。然后将混合物浓缩并用etoac稀释,用饱和na2co3洗涤,在na2so4上干燥,过滤并浓缩,产生作为黄色油状物的化合物cvie401(38mg,79%)。
[0351]
化合物53的合成:(5-((6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-17-氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)戊基)(甲基)氨基甲酸叔丁酯
[0352][0353]
在-78℃下,向n-boc-n-甲基-5-三苯基鏻戊烯胺碘化物(4.39g,7.45mmol)在thf(45ml)中的混合物逐滴添加正丁基锂在thf中的溶液(4.46ml,2.5n,11.16mmol)。然后将混合物在0℃搅拌20min。将混合物冷却至-50℃并添加化合物39(830mg,2.48mmol)。将混合物在室温搅拌过夜。将所述混合物用h2o淬灭,浓缩,并通过柱层析进行纯化(pe/etoac=1/1),然后通过制备hplc进行纯化,得到作为白色固体的化合物53(80mg,300mg)。
[0354]
cvie406的合成:(6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-3-(5-(甲
基)丁酸乙酯;4-(6α-羟基-17-酮基雄甾烷-3-基)己酸乙酯;4-(6β-羟基-17-酮基雄甾烷-3-基)己酸;(e,z)-3-(5-n-甲基氨基亚戊基]-6α-羟甲基雄甾烷-7,17-二酮;(e,z)-3-[2-(吡咯烷-3-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮;(e,z)-3-[2-(氮杂环丁烷-2-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮;(e,z)-3-[2-(哌啶-4-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮;(e,z)-3-(5-n-甲基氨基亚戊基)-6α-羟甲基-7α-羟基雄甾烷-17-酮;3β-[2-(氮杂环丁烷-2-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮;3β-[2-(氮杂环丁烷-2-基)乙基]-6α-羟甲基-7α-羟基雄甾烷-17-酮;3β-[2-(吡咯烷-3-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮;3β-[2-(吡咯烷-3-基)乙基]-6α-羟甲基-7α-羟基雄甾烷-17-酮;3β-[2-(哌啶-4-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮;和3β-[2-(哌啶-4-基)乙基]-6α-羟甲基-7α-羟基雄甾烷-17-酮。
[0371]
方面6:方面1所述的化合物,其选自:(e)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸;(z)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸;(e)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸;(z)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸;(6α-羟基-17-酮雄甾烷-3β-基)乙酸乙酯;4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸;4-(6β-羟基-17-酮基雄甾烷-3-基)丁酸;2-(6β-羟基-17-酮基雄甾烷-3-基)乙酸;4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸乙酯;4-(6α-羟基-17-酮基雄甾烷-3-基)己酸乙酯;和4-(6β-羟基-17-酮基雄甾烷-3-基)己酸。
[0372]
方面7:方面1所述的化合物,其选自4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸和2-(6β-羟基-17-酮基雄甾烷-3-基)乙酸。
[0373]
方面8:方面1-7中的任一者所述的化合物,其中所述可药用盐选自氯化物、溴化物、硫酸盐、磷酸盐、硝酸盐、延胡索酸盐、琥珀酸盐、草酸盐、苹果酸盐、酒石酸盐、马来酸盐、柠檬酸盐、甲磺酸盐和苯甲酸盐。
[0374]
方面9:一种药物组合物,其用于心力衰竭的治疗方法中,所述药物组合物包含一种或多种方面1-8中的任一者所述的化合物与至少一种可药用载体和/或赋形剂的组合。
[0375]
方面10:方面9所述的药物组合物,其被配制成用于肠内给药、肠胃外给药或吸入。
[0376]
方面11:方面10所述的药物组合物,其被配制成用于口服给药。
[0377]
方面12:方面11所述的药物组合物,其以约1mg/kg至约20mg/kg之间的剂量给药,任选地所述剂量在约1mg/kg至约10mg/kg之间。
[0378]
方面13:方面9所述的药物组合物,其被配制成用于静脉内注射。
[0379]
方面14:方面13所述的药物组合物,其以约0.125mg/kg至约10mg/kg之间的剂量给药,任选地所述剂量在约0.25mg/kg至约5mg/kg之间。
[0380]
方面15:方面9所述的药物组合物,其被配制成用于肌肉内注射。
[0381]
方面16:方面15所述的药物组合物,其以约0.25mg/kg至约50mg/kg之间的剂量给药,任选地所述剂量在约0.25mg/kg至约35mg/kg之间。
[0382]
方面17:方面9-16中的任一者所述的药物组合物,其每天至少给药一次。
[0383]
方面18:方面9-17中的任一者所述的药物组合物,其还包含一种或多种另外的治疗活性成分。
[0384]
方面19:方面18所述的药物组合物,其中所述一种或多种另外的治疗活性成分选自ace抑制剂、airb、利尿剂、ca
2
通道阻断剂、β-阻断剂、毛地黄、no供体、血管扩张剂、serca2a刺激剂、脑啡肽酶(nep)抑制剂、肌球蛋白丝激活剂、重组松弛素-2介导物、重组np
3-[2-(哌啶-4-基)亚乙基]-6α-羟基雄甾烷-17-酮;3β-[2-(哌啶-4-基)乙基]-6α-羟基雄甾烷-17-酮;(6α-羟基-17-酮雄甾烷-3β-基)乙酸乙酯;4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸;4-(6β-羟基-17-酮基雄甾烷-3-基)丁酸;2-(6β-羟基-17-酮基雄甾烷-3-基)乙酸;4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸乙酯;4-(6α-羟基-17-酮基雄甾烷-3-基)己酸乙酯;4-(6β-羟基-17-酮基雄甾烷-3-基)己酸;(e,z)-3-(5-n-甲基氨基亚戊基]-6α-羟甲基雄甾烷-7,17-二酮;(e,z)-3-[2-(吡咯烷-3-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮;(e,z)-3-[2-(氮杂环丁烷-2-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮;(e,z)-3-[2-(哌啶-4-基)亚乙基]-6α-羟甲基雄甾烷-7,17-二酮;(e,z)-3-(5-n-甲基氨基亚戊基)-6α-羟甲基-7α-羟基雄甾烷-17-酮;3β-[2-(氮杂环丁烷-2-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮;3β-[2-(氮杂环丁烷-2-基)乙基]-6α-羟甲基-7α-羟基雄甾烷-17-酮;3β-[2-(吡咯烷-3-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮;3β-[2-(吡咯烷-3-基)乙基]6α-羟甲基-7α-羟基雄甾烷-17-酮;3β-[2-(哌啶-4-基)乙基]-6α-羟甲基雄甾烷-7,17-二酮;和3β-[2-(哌啶-4-基)乙基]-6α-羟甲基-7α-羟基雄甾烷-17-酮。
[0405]
方面33:方面28所述的方法,其中所述显著纯serca2a刺激剂选自:(e)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸;(z)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸;(e)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸;(z)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸;(6α-羟基-17-酮雄甾烷-3β-基)乙酸乙酯;4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸;4-(6β-羟基-17-酮基雄甾烷-3-基)丁酸;2-(6β-羟基-17-酮基雄甾烷-3-基)乙酸;4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸乙酯;4-(6α-羟基-17-酮基雄甾烷-3-基)己酸乙酯;和4-(6β-羟基-17-酮基雄甾烷-3-基)己酸。
[0406]
方面34:方面28所述的方法,其中所述显著纯serca2a刺激剂选自4-(6α-羟基-17-酮基雄甾烷-3-基)丁酸和2-(6β-羟基-17-酮基雄甾烷-3-基)乙酸。
[0407]
方面35:方面28-34中的任一者所述的方法,其中所述可药用盐选自氯化物、溴化物、硫酸盐、磷酸盐、硝酸盐、延胡索酸盐、琥珀酸盐、草酸盐、苹果酸盐、酒石酸盐、马来酸盐、柠檬酸盐、甲磺酸盐和苯甲酸盐。
[0408]
方面36:方面28-35中的任一者所述的方法,其中所述药物组合物口服给药。
[0409]
方面37:方面36所述的方法,其中所述药物组合物以约1mg/kg至约20mg/kg之间的剂量给药,任选地所述剂量在约1mg/kg至约10mg/kg之间。
[0410]
方面38:方面28-35中的任一者所述的方法,其中所述药物组合物静脉内给药。
[0411]
方面39:方面38所述的方法,其中所述药物组合物以约0.125mg/kg至约10mg/kg之间的剂量给药,任选地所述剂量在约0.25mg/kg至约5mg/kg之间。
[0412]
方面40:方面28-35中的任一者所述的方法,其中所述药物组合物肌肉内给药。
[0413]
方面41:方面40所述的方法,其中所述药物组合物以约0.25mg/kg至约50mg/kg之间的剂量给药,任选地所述剂量在约0.25mg/kg至约35mg/kg之间。
[0414]
方面42:方面28-41中的任一者所述的方法,其中所述药物组合物包含一种或多种另外的治疗活性成分。
[0415]
方面43:方面42所述的方法,其中所述一种或多种另外的治疗活性成分选自ace抑制剂、airb、利尿剂、ca
2
通道阻断剂、β-阻断剂、毛地黄、no供体、血管扩张剂、serca2a刺激剂、脑啡肽酶(nep)抑制剂、肌球蛋白丝激活剂、重组松弛素-2介导物、重组np蛋白、可溶性
鸟苷酸环化酶(sgc)的激活剂和血管紧张肽ⅱ受体的β-抑制蛋白配体。
[0416]
方面44:方面43所述的方法,其中所述利尿剂选自呋塞米、布美他尼、托拉塞米、美托拉宗、醛固酮拮抗剂、噻嗪类利尿剂。
[0417]
方面45:方面43所述的方法,其中所述ace抑制剂是赖诺普利或雷米普利。
[0418]
方面46:方面42所述的方法,其中所述一种或多种另外的治疗活性成分选自缬沙坦、坎地沙坦、奥美沙坦、替米沙坦、氯沙坦、沙库比曲、卡维地洛、omecamtiv和美托洛尔。
[0419]
方面47:方面28-46中的任一者所述的方法,其中所述个体是人类。
[0420]
方面48:方面28-47中的任一者所述的方法,其中所述一种或多种心功能参数选自ca
2
瞬时(cat)幅度、ca
2
诱导的ca
2
释放(cicr)、90%复极化时的动作电位持续时间(apd90)的刺激率依赖性、舒张期膜电位(e
diast
)、最大去极化速度(dv/dt
max
)、心率、心脏压力、收缩压、舒张压、lvef、e/e’比率、e/ea比率、e/a比率和每搏输出量。
[0421]
方面49:方面28-48中的任一者所述的方法,其中所述测量步骤在所述给药步骤之前、期间和/或之后进行。
[0422]
下面的实施例进一步说明本发明。
[0423]
实施例1:式(i)的化合物的制备
[0424]
在下面的实施例中,除非另有陈述,否则化学化合物、溶剂、反应物和任何其他材料来自于商业化来源。通常,式(i)的化合物从脱氢表雄酮(普拉睾酮)开始通过多步合成来制备。脱氢表雄酮是商业化产品,或者可以从4-雄烯-3,17-二酮(雄烯二酮)开始按照公知的方法来制备。
[0425]
5α-雄甾烷-3β,6α,17β-三醇的制备
[0426][0427]
如全部内容通过引用并入本文的de munari等(j.med.chem.,2003,46(17):3644-54)中所描述,适用于合成6-α-3,17-雄甾烷二酮(2)的中间体从脱氢表雄酮1通过硼氢化和随后的氧化来生产。简单来说,将脱氢表雄酮1(5g,17.5mmol,1当量)在thf(85ml)中的溶液在-20℃和ar下搅拌。然后向所述搅拌的溶液添加thf中的1m bh3·
thf络合物(44ml,44mmol,2.5当量),并将搅拌在室温继续3小时。小心地逐滴添加h2o(85ml),然后逐滴添加nabo3·
4h2o(5.4g,35mmol,2当量)。在室温搅拌过夜后,将所述混合物过滤。将固体用thf洗涤,然后舍弃。将清液用nacl饱和并用thf萃取(3
×
40ml)。将合并的有机萃取液在nacl和na2so4上干燥、过滤并蒸发至干。将5α-雄甾烷-3β,6α,17β-三醇2粗产物从etoac/meoh(2/1,10ml/g)结晶,得到白色固体(3.8g,70%)。
[0428]
5α-雄甾烷-3β,6α,17β-三醇2的光谱数据:
[0429]1h nmr(dmso-d6)δ4.44(m,1h,oh),4.42(m,1h,oh),4.24(d,1h,oh),3.42(dt,1h,16-ha),3.26(m,1h,3-h),3.12(m,1h,6-h),0.72(s,3h,ch3),0.60(s,3h,ch3)。mp 232-234℃。
[0430]
6α-羟基雄甾烷-3,17-二酮的制备
[0431][0432]
中间体3从2通过c3和c17位置处的选择性氧化来获得。在0℃下向5α-雄甾烷-3β,6α,17β-三醇2(2g,6.5mmol,1当量)在二噁烷/h2o/吡啶(54/10/1ml)中的搅拌的溶液添加nbs(3.4g,19.5mmol,3当量)。在添加后,允许所述混合物升温至室温并搅拌过夜。将所述橙色溶液用水(50ml)稀释并用na2s2o3(350mg)淬灭。在真空下蒸发掉有机溶剂,直至白色固体出现。将所述固体过滤并用水洗涤。在40℃下干燥后,获得作为白色固体的6α-羟基雄甾烷-3,17-二酮3(1.3g,70%)。
[0433]
6α-羟基雄甾烷-3,17-二酮3的光谱数据:
[0434]1h nmr(丙酮-d6)δ3.61(d,1h,oh),3.48(m,1h,6-h),1.11(s,3h,ch3),0.86(s,3h,ch3)。mp 204-206℃,文献值为206-207(hammerschmidt&spiteller,1973)
[0435]
雄甾烷-3-亚甲基-17-酮的合成
[0436][0437]
然后通过在c3羰基上的选择性维蒂希反应和随后与5-戊烯酸的交叉复分解偶联,将6-α-3,17-雄甾烷二酮3转变成外甲烷衍生物6(雄甾烷-3-亚甲基-17-酮)。在-5℃下,向甲基三苯基溴化膦(1,66g,6mmol,4当量)在thf(10ml)中的悬液添加叔丁醇钾(670mg,6mmol,4当量)。所述溶液立即变色成亮橙色。10分钟后,添加6α-羟基雄甾烷-3,17-二酮3(450mg,1.5mmol,1当量),同时保持温度低于0℃。在添加后,立即通过添加1m hcl水溶液(15ml)将反应淬灭,并用etoac萃取(3x20ml)。将合并的有机相在na2so4上干燥并蒸发至干。将所述粗萃取物通过柱层析进行纯化(洗脱剂etoac:石油精4:6),产生376mg(83%)作为白色泡沫的雄甾烷-3-亚甲基-17-酮6。
[0438]
雄甾烷-3-亚甲基-17-酮6的光谱数据:
[0439]1h nmr(400mhz,氯仿-d)δ4.63(dt,2h,3α-ch2),3.47(td,1h,6-h),2.55(ddd,1h,16ha),2.44(ddd,1h,16-hb),0.89(s,3h,ch3),0.86(s,3h,ch3),0.80
–
0.70(m,1h,5-h)。
[0440]
13
c nmr(101mhz,氯仿-d)δ220.79(17-c),148.22(3-c),107.40(3-ch2),69.85(6-c),13.79(ch3),12.91(ch3)。
[0441]
通过交叉复分解反应从前体6直接合成cvie 201和202
[0442][0443]
向雄甾烷-3-亚甲基-17-酮6(100mg,0.33mmol,1当量)在dcm(1ml)中的溶液添加hoveyda-grubbs第二代催化剂(12mg,0.015mmol,0.05当量)。然后将所述溶液在回流下加
热,并且每20分钟用10μl 4-戊烯酸处理(总共330μl,3.3mmol,10当量)。在添加结束后,将混合物继续回流2h。将反应混合物真空浓缩并通过快速层析进行纯化(洗脱剂丙酮:石油精3:7 0.1%hco2h),以获得两种不同的白色固体(e)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸(4.8mg,4%)(cvie201)和(z)-4-(6α-羟基-17-酮基雄甾烷-3-亚基)丁酸(7.2mg,6%)(cvie202)。
[0444]
cvie201的光谱数据:
[0445]1h nmr(氯仿-d)δ5.10(t,j=7.3hz,1h,3α-h),3.50(td,j=10.3,9.8,6.0hz,1h,6-h),2.91(d,1h,16-ha),0.89(s,3h,ch3),0.86(s,3h,ch3),0.73(m,1h,5-h)。
[0446]
13
c nmr(101mhz,氯仿-d)δ221.30(17-c),178.72(co2),139.71(3-c),119.89(3α-c),69.95(6-c),54.03(5-c),24.03(16-c),13.93(ch3),13.06(ch3)。
[0447]
ms(esi):为c
23h33o4-[m-]计算的值为373,2。实测值:373.4
[0448]
cvie202的光谱数据:
[0449]1h nmr(400mhz,氯仿-d)δ5.10(t,1h,3α-h),3.50(td,1h,6-h),2.91(d,1h,16-ha),0.89(s,3h,ch3),0.86(s,3h,ch3),0.73(t,1h,5-h)。
[0450]
13
c nmr(101mhz,氯仿-d)δ220.86(17-c),177.66(co2),139.84(c-3),119.47(ca),70.08(c-6),13.78(ch3),12.91(ch3)。
[0451]
或者,通过修改维蒂希反应来获得cvie201和cvie202。在一种方法(路线a)中,利用极性溶剂例如dmso和碱例如nah使甜菜碱中间体稳定。第二种方法(路线b)允许使用非质子性溶剂例如thf作为碱使氧杂磷杂环丁烷中间体稳定。路线a产生非对映异构体的混合物(60%的z/顺式cvie202;30%的e/反式cvie201),而路线b提供源自于氧杂磷杂环丁烷中间体的cvie202。任一程序都需要产生如下所述的非对映异构体7和/或8。
[0452]
cvie 201和202通过维蒂希反应的可选合成
[0453]
路线a
[0454][0455]
在ar气氛下向无水dmso(1ml)小心地添加矿物油中的60%nah(100mg,2.56mmol,8当量)。将得到的溶液在60℃搅拌20分钟。在室温冷却后,添加(3-羧基丙基)三苯基溴化膦(550mg,1.28mmol,4当量)。立即出现亮橙色颜色。将溶液搅拌2h。然后向所述混合物添加6α-羟基雄甾烷-3,17-二酮3(100mg,0.32mmol,1当量)。允许得到的溶液在室温继续搅拌4h。将反应混合物用etoac(25ml)稀释,并用1m hcl水溶液(3x 30ml)洗涤。将有机层在na2so4上干燥并蒸发至干,得到25mg粗制品。
[0456]
将所述粗制品首先溶解在meoh(1.5ml)中,然后添加edc盐酸盐(115mg,0.6mmol,2当量)和dmap(5mg,0.03mmol,0.1当量)。将所述溶液在室温搅拌3h。在真空浓缩后,将粗品固体溶解在etoac(15ml)中并用1m hcl水溶液(3x 10ml)洗涤。将粗产物在硅胶上通过快速层析进行纯化(丙酮:石油精3:7),得到25mg包含非对映异构体7和8的混合物的透明油状物(20%)。
[0457]
两种非对映异构体7和8的光谱数据:
[0458]1h nmr(氯仿-d)δ5.16-4.96(m,1h,3α-h),3.66(s,3h,ch3o),3.47(m,1h,6-oh),0.89(s,3h,ch3),0.86(s,3h,ch3),0.74(m,1h,5-h)。
[0459]
路线b
[0460][0461]
在ar气氛和-40℃下,向(3-羧基丙基)三苯基溴化膦(8.5g,20mmol,6当量)的无水thf(33ml)悬液小心地添加thf中的1m lihmds溶液(40ml,40mmol,12当量)。将所述溶液在-40℃搅拌过夜,直至出现亮橙色。然后在-40℃下向所述溶液添加6α-羟基雄甾烷-3,17-二酮3(1g,3.3mmol,1当量)。在室温搅拌过夜后,将反应混合物用1m hcl水溶液(300ml)淬灭并用etoac(3x350ml)萃取。将合并的有机层在na2so4上干燥并蒸发至干。
[0462]
将所述粗制品溶解在无水etoh(17ml)中,然后添加edc盐酸盐(1.26mg,6.6mmol,2当量)和dmap(50mg,0.3mmol,0.1当量)。允许所述混合物在室温搅拌3h。将反应在etoac(150ml)中稀释并用1m hcl水溶液(3x 100ml)洗涤。将粗产物在硅胶上通过快速层析进行纯化(丙酮:石油精3:7)得到910mg(72%)化合物8。
[0463]
化合物8的光谱数据:
[0464]1h nmr(氯仿-d)δ5.07(t,1h,3α-h),4.10(q,2h,och2),3.47(td,1h,6-h),2.91(d,1h),0.88(s,3h,ch3),0.85(s,3h,ch3),0.71(m,1h,5-h)。
[0465]
13
c nmr(101mhz,氯仿-d)δ173.67(17-c),139.60(3α-c),119.94(3-c),69.98(6-c),60.41(och2),51.33(5-c),40.37(ch3),13.92(ch3),13.08(ch3)。
[0466]
甲基(或乙基)酯的最终水解
[0467][0468]
向甲基酯7和8(25mg,0.06mmol,1当量)在thf(600μl)和水(200μl)中的溶液添加1m lioh水性溶液(150μl,2.5当量)。2h后,将反应用水(10ml)稀释,并通过添加1m hcl直至溶液达到ph 1进行淬灭。将水性相用etoac(3x 15ml)萃取。将合并的有机层在na2so4上干燥并蒸发至干。将粗品通过快速层析进行纯化(acoet:石油精7:3 1%hcooh)。获得两种白色固体,对应于e(7mg,31%)和z(12mg,54%)非对映异构体(分别为cvie201和cvie202)。
[0469]1h nmr(氯仿-d)δ5.12(bt,1h,3α-h),3.51-3.42(m,1h,6-h),0.90(s,3h,ch3),0.86(s,3h,ch3),0.79-0.69(m,1h,5-h)。
[0470][0471]
cvie 203和204的合成方式:中间体化合物12的合成
[0472]
为了制备cvie203和cvie204,首先从6-α-3,17-雄甾烷二酮3生产前体12。通过与乙二醇和酸催化剂(p-tsa或樟脑磺酸)的组合在甲苯中反应,将6-α-3,17-雄甾烷二酮3的羰基作为二缩酮进行保护,得到化合物9。将化合物9用pcc或其他氧化剂氧化,得到化合物10,然后将其用nabh4或kbh4还原,产生c6-羟基选择性地为β-构型的被保护的醇11。如de munari等(j.med.chem.,2003,46(17):3644-54)中所述在丙酮中通过酸性处理对所述环状二缩酮进行最后切割,得到前体12。
[0473]
简单来说,将6α-羟基雄甾烷-3,17-二酮(1.5g,4.9mmol,1当量)、乙二醇(10.5ml,88mmol,36当量)和ptsa(561mg,2.9mmol,0.6当量)在甲苯(160ml)中的溶液使用dean-stark分水器在回流下搅拌12h。在冷却至室温后,将所述混合物用5%nahco3水溶液中和。分离有机层并用h2o(2
×
40ml)洗涤,在na2so4上干燥并蒸发至干,产生作为白色固体化合物的3,3:17,17-双(亚乙基二氧)雄甾烷-6α-醇9(1.9g,98%)。
[0474]
3,3:17,17-双(亚乙基二氧)雄甾烷-6α-醇9的光谱数据:
[0475]1h nmr(dmso-d6)δ4.25(d,1h,oh),3.88-3.70(m,8h,och2),3.11(m,1h,6-h),0.74(s,3h,ch3),0.73(s,3h,ch3)。
[0476]
在0℃下,向3,3:17,17-双(亚乙基二氧)雄甾烷-6α-醇(3g,14mmol,1当量)9和抗坏血酸钠(1.2g,14mmol,4当量)在无水ch2cl2(87ml)中的溶液添加pcc(148mg,0.69mmol,4当量)。将所述混合物在室温搅拌过夜。将混合物用1m hcl水溶液(3x 30ml)和水(3x30ml)洗涤。将有机层在na2so4上干燥并蒸发至干。将粗品在硅胶柱上通过快速层析进行纯化(洗脱剂丙酮:石油精2:8)。获得作为白色固体的3,3:17,17-双(亚乙基二氧)雄甾烷-6-酮10(1.53g(96%))。
[0477]
3,3:17,17-双(亚乙基二氧)雄甾烷-6-酮10的光谱数据:
[0478]1h nmr(丙酮-d6)δ3.97-3.76(m,8h,ch2o),2.19(dd,1h,16-ha),0.84(s,3h,ch3),0.75(s,3h,ch3)。
[0479]
在0℃下向3,3:17,17-双(亚乙基二氧)雄甾烷-6-酮10(1g,2.5mmol,1当量)在meoh(13ml)中的搅拌的悬液添加nabh4(144mg,3mmol,1.2当量)。在0℃下2h后,逐滴添加h2o(40ml)。将所述混合物用etoac(3
×
40ml)萃取。将合并的有机萃取液在na2so4上干燥、过滤并蒸发至干,以得到白色固体,其是3,3:17,17-双(亚乙基二氧)雄甾烷-6β-醇11(915mg,92%)。
[0480]
3,3:17,17-双(亚乙基二氧)雄甾烷-6β-醇11的光谱数据:
[0481]1h nmr(丙酮-d6)δ3.95-3.75(m,8h,och2),3.70(m,1h,6-h),3.33(d,1h,6-oh),
1.05(s,3h,ch3),0.84(s,3h,ch3)。
[0482]
向3,3:17,17-双(亚乙基二氧)雄甾烷-6β-醇11(910mg,2.3mmol,1当量)在丙酮(46ml)中的溶液在5分钟内添加分成小份的ptsa(2.26g,11.5mmol,5当量)。在室温搅拌1h后,通过添加5%nahco3水溶液直至ph 7,将所述溶液淬灭。在搅拌5分钟后出现白色固体。在真空中除去挥发物。将悬液用ch2cl2(3x 30ml)萃取,并将合并的有机萃取液用盐水(40ml)洗涤,在na2so4上干燥,过滤并蒸发。将得到的固体与正己烷/etoac 8/2(10ml)搅拌45分钟,然后通过过滤进行收集。将所述固体在45℃干燥3小时。得到568mg(81%)白色固体(即6β-羟基雄甾烷-3,17-二酮12)。
[0483]
6β-羟基雄甾烷-3,17-二酮12的光谱数据:
[0484]1h nmr(dmso-d6)δ4.47(d,1h,oh),3.57(m,1h,6-h),1.13(s,3h,ch3),0.81(s,3h,ch3)。
[0485]
12转变成最终的cvie 203和204
[0486][0487]
然后使用上文为cvie201和cvie202描述的相同程序,通过维蒂希反应从前体12获得cvie203和cvie204。利用noesy实验来鉴定两种异构体中c3-c1’双键处的构型。
[0488]
简单来说,在ar气氛下向无水dmso(1ml)小心地添加矿物油中的60%nah(100mg,2.56mmol,8当量)。将得到的溶液在60℃搅拌20分钟。在室温冷却后,添加(3-羧基丙基)三苯基溴化膦(550mg,1.28mmol,4当量)。立即出现亮橙色颜色。将所述溶液搅拌2h。然后向所述混合物添加6β-羟基雄甾烷-3,17-二酮12(100mg,0.32mmol,1当量)。允许得到的溶液在室温继续搅拌4h。将反应混合物用etoac(25ml)稀释并用1m hcl水溶液(3x 30ml)洗涤。将有机层在na2so4上干燥并蒸发至干,得到25mg粗制品。
[0489]
然后将所述粗制品溶解在meoh(1.5ml)中。添加edc盐酸盐(115mg,0.6mmol,2当量)和dmap(5mg,0.03mmol,0.1当量)。将所述溶液在室温搅拌3h。在真空浓缩后,将粗品固体溶解在etoac(15ml)中并用1m hcl水溶液(3x 10ml)洗涤。将所述粗产物在硅胶上通过快速层析进行纯化(丙酮:石油精3:7),得到非对映异构体13和14的混合物,得率分别为17%和30%。
[0490]
化合物13的光谱数据:
[0491]1h nmr(400mhz,氯仿-d)δ5.07(bs,1h,3α-h),3.85(d,1h,6-h),3.66(s,3h,och3),2.54
–
2.39(m,2h,3γ-h),1.10(s,3h,ch3),0.89(s,3h,ch3),0.80
–
0.69(m,1h,5-h)。
[0492]
13
c nmr(101mhz,氯仿-d)δ219.74(17-c),167.24(co2),140.38(3-c),119.60(3α-c),71.52(6-c),54.45(5-c),51.22(och3),14.09(ch3),13.86(ch3)。
[0493]
化合物14的光谱数据:
[0494]
1h nmr(400mhz,氯仿-d)δ5.07(s,1h,3α-h),3.89(d,1h,6-h),3.66(s,3h,och3),2.46(dd,1h,16-ha),1.10(s,3h,ch3),0.89(s,3h,ch3),0.79
–
0.64(m,1h,5-h)。
[0495]
13c nmr(101mhz,氯仿-d)δ221.21(17-c),176.10(co2),140.33(3-c),119.39(3α-c),71.75(6-c),54.49(5-c),51.20(och3)15.24(ch3),13.87(ch3)。
[0496]
将反应混合物真空浓缩并通过快速层析进行纯化(洗脱剂丙酮:石油精3:7 0.1%hco2h),得到两种不同的白色固体(e)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸(cvie203)和(z)-4-(6β-羟基-17-酮基雄甾烷-3-亚基)丁酸(cvie204)。
[0497]
化合物cvie203的光谱数据:
[0498]1h nmr(400mhz,丙酮-d6)δ4.84(bt,1h,3α-h),3.55(d,1h,6-h),0.90(s,3h,ch3),0.61(s,3h,ch3),0.51(ddd,1h,5-h)。
[0499]
13
c nmr(101mhz,丙酮-d6)δ218.86(17-c),173.42(co2),140.74(3-c),119.29(3α-c),70.18(6-c),14.74(ch3),13.18(ch3)。
[0500]
化合物cvie204的光谱数据:
[0501]1h nmr(400mhz,氯仿-d)δ5.07(s,1h,3α-h),3.88(s,1h,6-h),1.08(s,3h,ch3),0.88(s,3h,ch3),0.72(s,1h,5-h)。
[0502]
13
c nmr(101mhz,丙酮)δ216.39(17-c),173.80(co2),135.48(3-c),113.80(3α-c),66.52(6-c),10.07(ch3),8.70(ch3)。
[0503]
通过加氢和酯水解生产cvie214、cvie215和cvie217
[0504][0505]
化合物cvie217从上述非对映异构体7 8的混合物生产。简单来说,所述非对映异构体的c3-c1’双键的加氢在etoac中使用pd-c催化来进行。得到的化合物是cvie217。c3处形成的立体中心的构型通过noesy实验来鉴定。然后将化合物cvie217用thf中的1m lioh或naoh水解,以产生cvie214。同样地,将非对映异构体13 14在etoac中使用pd-c催化进行加氢,以产生酯化合物19,然后将其用thf中的1m lioh或naoh水解,以产生cvie215。
[0506]
化合物19的光谱数据:
[0507]1h nmr(400mhz,氯仿-d)δ3.83(bs,1h,6-h),3.66(bs,3h,och3),2.44(dd,1h,16-ha),2.28(t,2h,3γ-h),0.99(s,3h,ch3),0.88(s,3h,ch3),0.79
–
0.70(m,1h,5-h)。
[0508]
13
c nmr(101mhz,氯仿-d)δ221.43(17-c),174.25(co2),71.96(6-c),51.25(och3),15.74(ch3),13.86(ch3)。
[0509]
化合物cvie214的光谱数据:
[0510]1h nmr(400mhz,氯仿-d)δ3.43(bt,1h,6-h),2.44(dd,1h,16ha),2.31(t,2h,3γ-h),0.84(s,3h,ch3),0.77(s,3h,ch3)。
[0511]
13
c nmr(101mhz,氯仿-d)δ221.35(17-c),179.11(co2),69.90(6-c),13.78(ch3),13.37(ch3)。
[0512]
化合物cvie215的光谱数据:
[0513]1h nmr(400mhz,氯仿-d)δ3.85(s,1h,6-h),2.45(dd,1h,16-ha),2.34(t,2h,3γ-h),1.00(s,3h,ch3),0.89(s,3h,ch3),0.74(d,1h,5-h)。
[0514]
13
c nmr(101mhz,氯仿-d)δ221.50(17-c),179.04(co2h),72.03(6-c),15.76(ch3),13.87(ch3)。
[0515]
化合物cvie217的光谱数据:
[0516]1h nmr(400mhz,氯仿-d)δ4.09(q,2h,ch2o),3.40(td,1h,6-h),2.49
–
2.36(dd,1h,16-ha),2.24(t,2h,3δ-h),0.83(s,3h,ch3),0.76(s,3h,ch3)。
[0517]
13
c nmr(101mhz,氯仿-d)δ220.91(17-c),173.77(co2),69.63(6-c),60.14(ch2o),14.23(ch3),13.76(ch3),13.36(ch3)。
[0518]
通过维蒂希反应和随后的c=c加氢和酯水解生产cvie213和cvie216
[0519][0520]
化合物6-α-3,17-雄甾烷二酮3也被用作通过horner-emmons反应合成cvie213和cvie216的起始点。首先,在ar气氛和0℃下,向矿物油中的60%nah(1.3g,33mmol,5当量)在dmf(200ml)中的悬液小心地添加磷酰基乙酸三乙酯(6.5ml,33mmol,5当量)。将得到的溶液在室温升温并搅拌20分钟。然后在0℃下添加6-α-3,17-雄甾烷二酮3(2g,6.5mmol,1当量)。在室温搅拌过夜后,通过小心地添加h2o(100ml)将反应淬灭,并用et2o(3x 150ml)萃取。将合并的有机层在na2so4上干燥并真空蒸发。将粗品在硅胶柱上通过快速层析进行纯化(丙酮:石油精3:7),产生2.1g(86%)两种非对映异构体的透明油状混合物(化合物21)。
[0521]
非对映异构体化合物21的光谱数据:
[0522]1h nmr(氯仿-d)δ5.60(d,hz,1h,3α-h),4.08(q,2h,ch2o),3.45(dq,1h,6-h),2.41(dd,1h,16-ha),0.90(s,3h,ch3),0.82(s,3h,ch3),0.77-0.66(m,1h,5-h)。
[0523]
13
c nmr(101mhz,氯仿-d)δ220.85(17-c),166.81(co2),161.87(3-c),113.75(3α-c),69.41(6-c),13.76(ch3),13.00(ch3)。
[0524]
在ar气氛下,向非对映异构体化合物21(2g,5.3mmol,1当量)在etoac(200ml)中的脱气的溶液添加10%pd-c(700mg)。在三个真空/氢气循环后,允许反应在h2气氛下在室温搅拌过夜。在通过真空/ar循环除去氢气后,将反应混合物在上过滤。将过滤的溶液蒸发至干。不需纯化获得1.8g(90%)cvie213产物。用thf中的1m lioh或naoh进一步水解cvie213产生cvie216。
[0525]
化合物cvie213的光谱数据:
[0526]1h nmr(氯仿-d)δ4.12(q,2h,och2),3.38(td,1h,6-h),2.42(dd,1h,16-ha),2.27(t,2h,3α-ch2),0.82(s,3h,ch3),0.76(s,3h,ch3)。
[0527]
13
c nmr(101mhz,氯仿-d)δ221.03(17-c),172.93(co2),69.43(6-c),13.76(ch3),13.32(ch3)。
[0528]
化合物cvie216的光谱数据:
[0529]1h nmr(400mhz,氯仿-d)δ3.43(td,1h,6-h),2.45(dd,1h,16-ha),2.28(t,3α-h),0.85(s,3h,ch3),0.80(s,3h,ch3)。
[0530]
13
c nmr(101mhz,丙酮)δ220.44(17-c),174.27(co2),68.57(6-c),12.99(ch3),12.59(ch3)。
[0531]
通过维蒂希反应和随后的c=c加氢和酯水解生产cvie218和cvie219
[0532][0533]
同样地,将6-α-3,17-雄甾烷二酮3与适合的三苯基鏻盐(例如5-羧基三苯基溴化膦、lihmds、thf,然后是etoh(或meoh))反应,产生化合物24。接下来,在氢气存在下使用pd-c催化将化合物24催化加氢产生cvie218,其在c-3位置处包括c6链。用thf中的1m lioh或naoh将cvie218水解产生cvie219。
[0534]
化合物cvie218的光谱数据:
[0535]1h nmr(400mhz,氯仿-d)δ4.15
–
4.05(m,2h,och2),3.40(td,1h,6-h),2.43(dd,1h,16-ha),2.26(td,2h,3ε-h),0.84(s,3h,ch3),0.76(s,3h,ch3)。
[0536]
13
c nmr(101mhz,氯仿-d)δ220.91(17-c),173.87(co2),69.81(6-c),60.14(ch2o),14.24(ch3),13.79(ch3),13.39(ch3)。
[0537]
化合物cvie219的光谱数据:
[0538]1h nmr(400mhz,氯仿-d)δ3.47
–
3.39(bt,1h,6-h),2.44(dd,1h,17-ha),2.33(t,2h,3ε-h),0.85(s,3h,ch3),0.77(s,3h,ch3)。
[0539]
13
c nmr(101mhz,氯仿-d)δ221.06(17-c),178.93(co2),69.91(6-c),13.80(ch3),13.39(ch3)。
[0540]
通过使用boc保护的胺的复分解反应和随后的boc去保护从前体6合成具有伯胺基的衍生物
[0541][0542]
对于具有伯胺基作为式(i)中的x取代基的衍生物的合成来说,使用上文为cvie201和cvie202的合成所述的相同的实验条件对前体6进行交叉复分解反应。
[0543]
简单来说,向雄甾烷-3-亚甲基-17-酮6在dcm中的溶液添加hoveyda-grubbs第二代催化剂。然后使用适合的boc保护的胺(例如叔丁基戊-4-烯-1-基氨基甲酸酯或n-boc-4-戊炔-1-胺)将雄甾烷-3-亚甲基-17-酮6与外亚甲基合并,产生非对映异构体25(25%得率)。将化合物25(50mg,0,1mmol,1当量)用500μl tfa/dcm(dcm中的三氟乙酸)的1:1混合物处理,然后在室温搅拌,以直接切割boc基团。在室温搅拌1分钟后,将反应用etoac(50ml)稀释并用饱和nahco3水溶液(3x30ml)洗涤。将有机相在na2so4上干燥,过滤并蒸发至干,产生作为白色固体的(ez)-3-(4-氨基亚丁基]-6α-羟基雄甾烷-17-酮(cvie209)(28mg,75%)。
[0544]
非对映异构体混合物25的光谱数据:
[0545]
1h nmr(400mhz,氯仿-d)δ5.18
–
5.03(m,1h,3α-h),3.46(td,1h,6-h),1.44(s,9h,t-bu),0.90(d,j=1.8hz,3h,ch3),0.86(s,3h,ch3),0.74(m,1h,5-h)。
[0546]
cvie209的光谱数据:
[0547]1h nmr(400mhz,cd3od)δ5.13(d,1h,3α-h),3.40(tt,1h,6-h),3.35
–
3.28(m,2h,3γ-h),0.96(s,3h,ch3),0.87(s,3h,ch3),0.83
–
0.70(m,1h,5-h)。
[0548]
13
c nmr(101mhz,cd3od)δ224.41(17-c),143.00(3a-c),142.68(3αb-c),124.31(3αa-c),124.02(3αb-c),72.75(c-6),16.70(ch3),15.78(ch3)。
[0549]
可选地,将化合物25与三甲基甲硅烷基碘化物在醇性溶剂(例如meoh)中反应,导致boc切割并伴有环外双键的迁移,以产生在c2与c3之间具有环内双键的cvie207。简单来说,在室温下向非对映异构体25(50mg,0,1mmol,1当量)的溶液添加dcm中的1m tmsi(100μl,0,1mmol,1当量)。在相同温度下搅拌2h后,在真空中除去溶剂。向残留物添加甲醇(2ml)并在室温放置1h。在真空中除去溶剂后,不需纯化获得cvie207。
[0550]
cvie207的光谱数据:
[0551]1h nmr(400mhz,cd3od)δ5.36(d,1h,h-2),3.44(td,1h,6-h),2.94(t,2h,3γ-h),2.45(dd,1h,16-ha),0.88(s,3h,ch3),0.79(s,3h,ch3)。
[0552]
13
c nmr(101mhz,cd3od)δ222.35(17-c),134.89(3-c),119.69(2-c),70.31(6-h),12.75(ch3),12.07(ch3)。
[0553]
具有环外不饱和性的环胺衍生物的合成:cvie205、cvie206、cvie210和cvie211
[0554][0555][0556]
环胺衍生物通过如上为cvie203和cvie204所述的氢化钠(nah)-dmso维蒂希反应来合成,并在同时利用适合的n-保护的鏻盐例如n-boc-4-(2-三苯基鏻乙基)氮杂环丁烷碘化物以产生化合物26和27,或利用n-boc-3-(2-三苯基鏻乙基)哌啶碘化物以产生化合物28和29。在纯化所述非对映异构体混合物后,通过用tfa酸水解切掉n-boc基团,以产生cvie205、cvie206、cvie210和cvie211。
[0557]
具有内环不饱和性的cvie208的合成(boc去保护期间的c=c双键迁移)
[0558][0559]
如上为cvie207的合成所述用tmsi进一步处理化合物28和29产生cvie208。
[0560]
加氢和使用tfa的boc切割以产生cvie212
[0561][0562]
可选地,将化合物28和29的双键催化加氢(h2,pd-c,etoac)以合成化合物30,然后在dcm中用tfa进行boc切割产生cvie212。
[0563]
带有6α-羟甲基雄甾烷-7,17-二酮的化合物的合成从共同中间体37开始来实现。化合物37本身从4-雄烯-3,17-二酮31开始,通过将两个酮组成部分用环状缩醛保护32并同时迁移双键,用重铬酸钠氧化烯丙基位置33,形成甲硅烷基烯醇醚35,用me3al和甲醛羟甲基化(36),以及最终在酸性条件下切割缩醛来合成。所述合成在下面的段落中更详细地描述。
[0564]
化合物32的合成:(20s,7r)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-12-烯
[0565][0566]
将雄甾-4-烯-3,17-二酮31(400.0g,1.4mol)和ptsa
·
h2o(13.3g,70.0mmol)在乙二醇(8.0l)中的混合物在100℃下搅拌,直至反应透亮。将约5.0l二醇在真空下蒸馏,以使沸腾温度在80-85℃左右。将所述混合物冷却到室温。将混合物调整到ph~9。然后将混合物倾倒在冰水中。将所述混合物过滤,将固体用水洗涤、收集并用丙酮研磨,得到作为黄色固体的粗品化合物32(469.0g,89%)。
[0567]
化合物33的合成:(20s,7r)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-12-烯-14-酮
[0568][0569]
将化合物32(440.0g,1.2mol)、hosu(541.2g,4.7mol)和na2cr2o7·
h2o(527.5g,1.8mol)在丙酮(8.0l)中的混合物在50℃剧烈搅拌2天。在冷却到室温后,将混合物用na2so3水溶液淬灭并搅拌20min。将混合物倾倒在冰水中。将得到的混合物搅拌20min,然后过滤。
将固体用水洗涤、收集并在真空中干燥,得到作为黄色固体的粗品化合物33(390.0g,85%)。
[0570]
化合物34的合成:(7s,20s)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-14-酮
[0571][0572]
将化合物33(50.0g,128.9mmol)在etoac(1250ml)中的混合物添加到pd/c(16.0g)。然后将所述混合物在h2下在室温搅拌过夜。tlc显示反应完成。将混合物过滤,浓缩,并通过快速层析进行纯化(pe/ea=2/1),得到作为白色固体的化合物34(25.0g,50.0%)。
[0573]
(7s,20s)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-14-酮34的光谱数据:
[0574]1h nmr(400mhz,dmso-d6):δ3.85-3.75(m,8h),2.44-2.35(m,2h),2.08-2.03(m,1h),1.87-1.79(m,2h),1.70-1.49(m,8h),1.41-1.28(m,4h),1.17-1.10(m,2h),1.03(s,3h),1.00-0.97(m,1h),0.76(s,3h)。
[0575]
化合物35的合成:1-((20s,7r)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-13-烯-14-基氧基)-1,1-二甲基-1-硅乙烷
[0576][0577]
将化合物34(20.0g,51.3mmol)在无水thf(100.0ml)中的混合物在-78℃下搅拌,然后逐滴添加甲苯中的1.5m lda(205.2ml,307.8mmol)。在相同温度下搅拌1小时后,逐滴添加me3sicl(50.0ml,400.1mmol)。在-70℃搅拌3小时后,将温度升高到-30℃并添加三乙胺(33.5g,331.5mmol)。在相同温度下搅拌1小时后,将混合物升温至室温并添加水(200.0ml)和etoac(100.0ml)。将分离的水性层用etoac萃取。将合并的有机层用盐水洗涤,在na2so4上干燥,过滤并蒸发至干。将残留物通过快速层析进行纯化(pe/ea=2/1),得到作为白色固体的化合物35(14.3g,60.3%)。
[0578]
化合物36的合成:(13s,20s,7r)-13-(羟甲基)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-14-酮
[0579][0580]
将2,6-二苯基酚(10.0g,27.6mmol)在无水dcm(450.0ml)中的混合物逐滴添加到
me3al在甲苯中的溶液(41.4ml,82.9mmol),同时用冰/水浴冷却,使得温度不超过室温。在室温搅拌1小时后,将所述溶液在0℃冷却,并逐滴添加三氧杂环己烷(24.8g,276.0mmol)在无水dcm(100.0ml)中的溶液。将所述浅黄色溶液在0℃继续搅拌1小时,然后将温度冷却至-78℃。添加化合物35(10.0g,27.6mmol)在无水dcm(125ml)中的溶液。在-78℃搅拌1h后,将温度升高到-20℃,并将反应混合物在该温度下搅拌过夜。在室温下添加5%nahco3水溶液(85.0ml)。将所述果冻状混合物通过垫过滤,用dcm充分洗涤。将分离的有机层用水洗涤并蒸发。向残留物添加thf中的约1m tbaf(24.0ml),并将所述溶液在室温搅拌1.5h。将所述溶液用水洗涤,在na2so4上干燥,过滤并蒸发至干。将残留物通过快速层析进行纯化,得到作为黄色固体的化合物36(6.5g,71.4%)。
[0581]
化合物37的合成:(6s,10r,13s)-6-(羟甲基)-10,13-二甲基十氢-1h-环戊烷并[a]菲-3,7,17(2h,4h,8h)-三酮
[0582][0583]
将化合物36(8.0g,19.0mmol)在丙酮(100.0ml)中的混合物添加到10%hcl水溶液(50.0ml)。然后将所述混合物加热到70℃1h。tlc显示反应完成。将混合物用5%naoh水溶液淬灭,并用dcm(50.0ml
×
2)萃取。将合并的有机相用盐水(50.0ml)洗涤,在na2so4上干燥,过滤,浓缩,并通过快速层析进行纯化(dcm/ea=4/1),得到粗产物,将其用乙醚研磨,得到作为白色固体的纯产物37(3.3g,52.4%)。
[0584]
(6s,10r,13s)-6-(羟甲基)-10,13-二甲基十氢-1h-环戊烷并[a]菲-3,7,17(2h,4h,8h)-三酮37的光谱数据:
[0585]1h nmr(400mhz,dmso-d6):δ4.14(t,1h),3.63-3.59(m,1h),3.50-3.47(m,1h),2.74-2.69(m,1h),2.42-2.27(m,5h),2.15-1.93(m,3h),1.87-1.79(m,1h),1.68-1.59(m,3h),1.54-1.44(m,2h),1.32-1.06(m,7h),0.81(s,3h)。
[0586]
lcms[流动相:在6.0min内从55%水(0.05%fa)和45%ch3cn(0.05%fa)到55%水(0.05%fa)和45%ch3cn(0.05%fa),最后在这些条件下0.5min],纯度》90%,rt=2.514min;ms计算值:332.2;ms实测值:333.2[m 1]
。
[0587]
化合物38的合成:(13s,14s,20s,7r)-13-(羟甲基)-7,20-二甲基二螺[1,3-二氧杂环戊烷-2,5'-四环[8.7.0.0《2,7》.0《11,15》]十七烷-14',2
”‑
1,3-二氧杂环戊烷]-14-醇
[0588][0589]
在0℃下向化合物36(22.0g,52.4mmol)在meoh(1000.0ml)中的混合物缓慢添加nabh4(4.0g,104.8mmol)。然后将所述混合物在室温搅拌1h。tlc显示反应完成。将混合物用5%nah2po4水溶液(220.0ml)淬灭,并用dcm(300.0ml
×
3)萃取。将合并的有机相用盐水
(200.0ml)洗涤,在na2so4上干燥,过滤,浓缩,并通过快速层析进行纯化(dcm/ea=4/1),得到粗产物,将其用乙醚研磨,得到作为白色固体的化合物38(7.5g,34.1%)。
[0590]
化合物39的合成:(8s,9s,15s,2r)-9-羟基-8-(羟甲基)-2,15-二甲基四环[8.7.0.0《2,7》.0《11,15》]十七烷-5,14-二酮
[0591][0592]
向化合物38(5.7g,13.5mmol)在丙酮(70.0ml)中的混合物添加10%hcl水溶液(35.0ml)。然后,将所述混合物加热到70℃1h。tlc显示反应完成。将所述混合物用5%naoh水溶液淬灭,并用dcm(50.0ml
×
2)萃取。将合并的有机相用盐水(50ml)洗涤,在na2so4上干燥,过滤并浓缩。将残留物通过快速层析进行纯化(dcm/ea=4/1),得到粗产物,将其用乙醚研磨,得到作为白色固体的纯产物39(1.8g,40.0%)。
[0593]
(8s,9s,15s,2r)-9-羟基-8-(羟甲基)-2,15-二甲基四环[8.7.0.0《2,7》.0《11,15》]十七烷-5,14-二酮39的光谱数据:
[0594]1h nmr(400mhz,dmso-d6):δ4.37(brs,1h),4.27(d,j=4.8hz,1h),3.87-3.86(m,1h),3.44-3.42(m,2h),2.45-1.87(m,10h),1.63-1.23(m,9h),0.99(s,3h),0.81(s,3h)。
[0595]
lcms[流动相:在6.0min内从55%水(0.05%fa)和45%ch3cn(0.05%fa)到55%水(0.05%fa)和45%ch3cn(0.05%fa),最后在这些条件下0.5min],纯度》90%,rt=2.515min;ms计算值:334.2;ms实测值:352.2[m 18]
。
[0596]
化合物40的合成:(2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)氮杂环丁烷-1-甲酸叔丁酯
[0597][0598]
在-78℃下向鏻盐(2.57g,4.5mmol)在thf(25ml)中的溶液添加正丁基锂在thf中的溶液(2.5m,3.6ml,9.0mmol)。将所述混合物在30℃搅拌1小时。接下来,在-20℃下向所述混合物添加化合物37(500mg,1.5mmol),然后升温至30℃2小时。将所述混合物用饱和nh4cl(25ml)淬灭,并用etoac(25ml
×
3)萃取。将合并的有机层浓缩,并将残留物在硅胶上通过柱层析进行纯化(己烷/etoac=1/1),得到粗品化合物。将所述化合物通过反相柱进行纯化,得到作为白色固体的纯化合物40(60mg,8%)。
[0599]
化合物40的数据:
[0600]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikwa diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=1.820min;ms计算值:499,ms实测值:400[m h-boc]
。
[0601]
化合物41的合成:3-(2-((3s,6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二
氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)氮杂环丁烷-1-甲酸叔丁酯
[0602][0603]
向化合物40(60mg,0.12mmol)在ea(3ml)中的溶液添加pd/c(60mg)。然后将所述混合物在h2下在室温搅拌过夜。将混合物过滤并将滤液浓缩,产生作为白色固体的化合物41(52mg,86%)。
[0604]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikma diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=1.911min;ms计算值:501,ms实测值:402[m h-boc]
。
[0605]
cvie407的合成:(3s,6s,10r,13s)-3-(2-(氮杂环丁烷-3-基)乙基)-6-(羟甲基)-10,13-二甲基十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0606][0607]
将化合物41(52mg,0.10mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌1小时。将所述混合物用饱和nahco3稀释,以调整到ph 8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,产生作为白色固体的化合物cvie407(13mg,32%)。
[0608]
cvie407的光谱数据:
[0609]1h nmr(cd3od,400mhz):δ3.86-3.82(m,2h),3.71-3.67(m,1h),3.54-3.47(m,2h),2.78-2.67(m,2h),2.56-2.39(m,3h),2.15-2.06(m,1h),1.85-1.50(m,12h),1.21-1.14(m,9h),1.07-1.01(m,2h),0.88(s,3h)。
[0610]
lcms柱:c18;柱尺寸:4.6
×
50mm;流动相:b(acn):a(0.02%nh4ac);梯度:6.5min内的(b%)-5-95-pos;rt=3.114min;ms计算值:401,ms实测值:402[m h]
。
[0611]
化合物42的合成:3-((e)-2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)氮杂环丁烷-1-甲酸叔丁酯
[0612][0613]
在-78℃下向化合物鏻盐(514mg,0.90mmol)在thf(5ml)中的溶液添加正丁基锂在thf中的溶液(2.5m,0.7ml,1.80mmol)。将所述混合物在40℃搅拌1小时。然后,在0℃下向所述混合物添加化合物39(100mg,0.30mmol),然后升温至40℃过夜。将反应重复9次。将混合物用饱和nh4cl(80ml)淬灭,并用etoac(100ml
×
3)萃取。将合并的有机层浓缩,并将残留物
通过制备hplc进行纯化,产生作为黄色固体的化合物42(20mg,1%)。
[0614]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikwa diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=1.945min;ms计算值:501,ms实测值:402[m h-boc]
。
[0615]
cvie403的合成:(6s,10r,13s)-3-(2-(氮杂环丁烷-3-基)亚乙基)-6-(羟甲基)-10,13-二甲基十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0616][0617]
将化合物40(130mg,0.26mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌1小时。将所述混合物用饱和nahco3碱化至ph8-9。将混合物用dcm(30ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie403(13mg,得率13%)。
[0618]
cvie403的光谱数据:
[0619]1h nmr(cd3od,400mhz):δ5.00-4.96(m,1h),3.92-3.81(m,3h),3.66-3.62(m,1h),3.54-3.52(m,2h),2.77-2.73(m,1h),2.67-2.61(m,2h),2.49-2.43(m,2h),2.38-2.29(m,2h),2.23-2.18(m,2h),2.06-1.96(m,2h),1.83-1.76(m,2h),1.71-1.61(m,3h),1.51-1.35(m,3h),1.18(s,3h),1.12-1.05(m,2h),0.98-0.90(m,1h),0.80(s,3h)。
[0620]
lcms柱:rt=3.964min;ms计算值:399,ms实测值:400[m h] 。
[0621]
化合物43的合成:3-(2-((6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-17-氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)氮杂环丁烷-1-甲酸酯
[0622][0623]
将化合物42(20mg,0.04mmol)、pd/c(10%,20mg)和pd(oh)2(20%,20mg)在etoac(2ml)中的混合物在h2下(在气球中)在室温搅拌过夜。将混合物过滤并将滤液浓缩,得到作为棕色固体的粗品化合物43(20mg,100%)。
[0624]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikma diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=1.978min;ms计算值:503,ms实测值:404[m h-boc]
。
[0625]
cvie408的合成:(6s,7s,10r,13s)-3-(2-(氮杂环丁烷-3-基)乙基)-7-羟基-6-(羟甲基)-10,13-二甲基十四氢-1h-环戊烷并[a]菲-17(2h)-酮
[0626]
[0627]
将化合物43(20mg,0.04mmol)在tfa/dcm(1:1,2ml)中的混合物在0℃搅拌30分钟。将所述混合物用饱和nahco3稀释,以调节到ph 8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie408(6.4mg,40%)。
[0628]
cvie408的光谱数据:
[0629]1h nmr(cd3od,400mhz):δ4.08(s,1h),3.84(t,j=8.4hz,1h),3.77-3.68(m,2h),3.52-3.48(m,2h),2.81-2.73(m,1h),2.52-2.44(m,1h),2.18-2.06(m,2h),1.86-1.71(m,4h),1.69-1.59(m,6h),1.52-1.43(m,2h),1.39-1.30(m,4h),1.24-1.15(m,4h),1.12-1.04(m,1h),0.95-0.92(m,1h),0.90(s,3h),0.87(s,3h)。
[0630]
lcms柱:柱:c18;柱尺寸:4.6
×
50mm;流动相:b(acn):a(0.02%nh4ac);梯度:6.5min内的(b%)-5-95-pos;rt=3.078min;ms计算值:403,ms实测值:404[m h]
。
[0631]
化合物44的合成:3-(2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)吡咯烷-1-甲酸叔丁酯
[0632][0633]
在-78℃下向化合物鏻盐(1.5g,2.62mmol)在thf(15ml)中的溶液添加正丁基锂在thf中的溶液(2.5m,1.57ml,3.94mmol)。将所述反应混合物在35℃搅拌1小时。然后,在-20℃下向所述混合物添加化合物37(350mg,1.05mmol)的溶液,并升温至室温2小时。将混合物用饱和nh4cl(25ml)淬灭,并用etoac(25ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过快速层析进行纯化(己烷:ea=1:1),得到粗品化合物。然后将所述化合物通过反相柱进行纯化,得到作为白色固体的纯化合物44(53mg,10%)。
[0634]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikma diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=2.017min;ms计算值:513,ms实测值:414[m h-boc]
。
[0635]
cvie402的合成:(6s,10r,13s)-6-(羟甲基)-10,13-二甲基-3-(2-(吡咯烷-3-基)亚乙基)十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0636][0637]
将化合物44(89mg,0.173mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌1小时。将所述混合物用饱和nahco3稀释,以调整到ph=8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,产生作为白色固体的化合物cvie402(38mg,53%)。
[0638]
cvie402的光谱数据:
[0639]1h nmr(cd3od,400mhz):δ5.08-5.05(m,1h),3.88-3.82(m,1h),3.63-3.59(m,1h),3.12-3.02(m,2h),2.99-2.92(m,1h),2.66-2.55(m,3h),2.48-2.42(m,2h),2.37-2.30(m,1h),2.23-2.19(m,1h),2.15-2.04(m,2h),2.03-1.91(m,4h),1.83-1.78(m,2h),1.75-1.61(m,3h),1.51-1.34(m,4h),1.19-1.17(m,3h),1.12-1.03(m,2h),0.97-0.90(m,1h),0.80(s,3h)。
[0640]
lcms柱:rt=3.060min;ms计算值:413,ms实测值:414[m h] 。
[0641]
化合物45的合成:3-(2-((3s,6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)吡咯烷-1-甲酸叔丁酯
[0642][0643]
将化合物44(53mg,0.103mmol)在ea(3ml)中的溶液添加到pd/c(60mg)。然后将所述混合物在h2下在室温搅拌过夜。将混合物过滤并将滤液浓缩,产生作为白色固体的化合物45(50mg,94%)。
[0644]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikma diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=1.984min;ms计算值:515,ms实测值:416[m h-boc]
。
[0645]
cvie409的合成:(3s,6s,10r,13s)-6-(羟甲基)-10,13-二甲基-3-(2-(吡咯烷-3-基)乙基)十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0646][0647]
将化合物45(50mg,0.09mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌1小时。将所述混合物用饱和nahco3稀释,以调整到ph 8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,产生作为白色固体的化合物cvie409(12mg,32%)。
[0648]
cvie409的光谱数据:
[0649]1h nmr(cd3od,400mhz):δ3.87-3.82(m,1h),3.70-3.64(m,1h),3.28-3.24(m,1h),3.21-3.16(m,1h),3.11-3.04(m,1h),2.72-2.62(m,2h),2.57-2.51(m,1h),2.47-2.39(m,2h),2.17-2.05(m,3h),1.85-1.70(m,5h),1.66-1.49(m,8h),1.38-1.34(m,2h),1.25-1.19(m,6h),1.14-1.10(m,2h),0.88(s,3h)。
[0650]
lcms柱:柱:c18;柱尺寸:4.6
×
50mm;流动相:b(acn):a(0.02%nh4ac);梯度:6.5min内的(b%)-5-95-pos;rt=3.180min;ms计算值:415,ms实测值:416[m h]
[0651]
化合物46的合成:3-(2-((6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-17-氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)吡咯烷-1-甲酸叔丁酯
[0652][0653]
在-78℃下向化合物鏻盐(527mg,0.90mmol)在thf(5ml)中的溶液添加正丁基锂在thf中的溶液(2.5m,0.7ml,1.80mmol)。将所述反应混合物在35℃搅拌1小时。然后在0℃下向所述混合物添加化合物39(100mg,0.30mmol),然后升温至35℃过夜。将反应重复四次。将所述混合物用饱和nh4cl(80ml)淬灭并用etoac(100ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,得到作为白色固体的化合物46(26mg,3%)。
[0654]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikma diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=2.000min;ms计算值:515,ms实测值:416[m h-boc]
。
[0655]
化合物47的合成:3-(2-((6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-17-氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)吡咯烷-1-甲酸叔丁酯
[0656][0657]
将化合物46(26mg,0.05mmol)、pd/c(10%,30mg)和pd(oh)2(20%,30mg)在etoac(3ml)中的混合物在h2下(在气球中)在室温搅拌过夜。将混合物过滤并将滤液浓缩,得到作为黄色固体的粗品化合物47(26mg,100%)。
[0658]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikma diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=2.059min;ms计算值:517,ms实测值:418[m h-boc]
。
[0659]
cvie410的合成:(6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-3-(2-(吡咯烷-3-基)乙基)十四氢-1h-环戊烷并[a]菲-17(2h)-酮
[0660][0661]
将化合物47(26mg,0.05mmol)在tfa/dcm(1:2,2ml)中的溶液在0℃搅拌1小时。将所述混合物用饱和nahco3稀释,以调整到ph 8-9。将混合物用dcm(20ml
×
3)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie410(9mg,43%)。
[0662]
cvie410的光谱数据:
[0663]1h nmr(cd3od,400mhz):δ3.95(s,1h),3.64-3.57(m,2h),3.12-3.07(m,1h),3.04-2.96(m,1h),2.93-2.89(m,1h),2.48-2.44(m,1h),2.38-2.32(m,1h),2.05-1.93(m,4h),1.69-1.57(m,5h),1.55-1.46(m,4h),1.37-1.33(m,4h),1.25-1.18(m,5h),1.08-0.90(m,3h),0.82-0.80(m,1h),0.77(s,3h),0.75(s,3h)。
[0664]
lcms柱:柱:c18;柱尺寸:4.6
×
50mm;流动相:b(acn):a(0.02%nh4ac);梯度:6.5min内的(b%)-5-95-pos;rt=3.139min;ms计算值:417,ms实测值:418[m h]
。
[0665]
化合物48的合成:4-(2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)哌啶-1-甲酸叔丁酯
[0666][0667]
在-78℃下向化合物鏻盐(2.16g,3.60mmol)在thf(16ml)中的混合物添加正丁基锂在thf中的溶液(2.5m,2.90ml,7.20mmol)。将所述反应混合物在30℃搅拌1小时。然后在-20℃下向所述混合物添加化合物37(400mg,1.20mmol)。将混合物在-20℃搅拌30分钟,然后升温至30℃2小时。将混合物用饱和nh4cl(15ml)淬灭并用etoac(30ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,得到作为黄色固体的化合物48(28mg,4%)。
[0668]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikma diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-30-95-pos;流速1.5ml/min,停止时间3min。rt=2.013min;ms计算值:527,ms实测值:428[m h-boc]
。
[0669]
cvie405的合成:(6s,10r,13s)-6-(羟甲基)-10,13-二甲基-3-(2-(哌啶-4-基)亚乙基)十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0670][0671]
将化合物46(80mg,0.152mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌30分钟。将所述混合物用饱和nahco3碱化至ph=8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie405(30mg,46%)。
[0672]
cvie405的光谱数据:
[0673]1h nmr(cd3od,400mhz):δ5.14(t,j=7.2hz,1h),3.88-3.80(m,1h),3.74-3.66(m,1h),3.09-3.06(m,2h),2.67-2.49(m,6h),2.45-2.38(m,1h),2.33-2.26(m,1h),2.19-2.04(m,2h),2.01-1.83(m,3h),1.80-1.69(m,6h),1.61-1.47(m,2h),1.45-1.34(m,2h),1.30-1.25(m,5h),1.19-1.08(m,4h),1.04-0.90(m,1h),0.88(s,3h)。
[0674]
lcms柱:rt=3.219min;ms计算值:427,ms实测值:428[m h] 。化合物49的合成:4-(2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二
[0675]
氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)哌啶-1-甲酸叔丁酯
[0676][0677]
将化合物48(28mg,0.05mmol)和pd/c(10%,50mg)在etoac(2ml)中的混合物在h2下(在气球中)在室温搅拌过夜。将混合物过滤并将滤液浓缩,得到作为黄色固体的粗品化合物49(28mg,100%)。
[0678]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikwa diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=2.109min;ms计算值:529,ms实测值:430[m h-boc]
。
[0679]
cvie411的合成:(6s,10r,13s)-6-(羟甲基)-10,13-二甲基-3-(2-(哌啶-4-基)乙基)十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0680][0681]
将化合物49(28mg,0.05mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌30分钟。将所述混合物用饱和nahco3稀释,以调整到ph 8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie411(10mg,43%)。
[0682]
cvie411的光谱数据:
[0683]1h nmr(cd3od,400mhz):δ3.85-3.81(m,1h),3.71-3.66(m,1h),3.07-3.04(m,2h),2.69(t,j=11.2hz,1h),2.63-2.51(m,3h),2.48-2.39(m,2h),2.15-2.05(m,1h),1.84-1.72(m,7h),1.63-1.46(m,4h),1.37-1.33(m,6h),1.28-1.14(m,7h),1.09-0.98(m,3h),0.88(s,3h)。
[0684]
lcms柱:柱:c18;柱尺寸:4.6
×
50mm;流动相:b(acn):a(0.02%nh4ac);梯度:6.5min内的(b%)-5-95-pos;rt=4.188min;ms计算值:429,ms实测值:430[m h]
。
[0685]
化合物50的合成:4-(2-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)乙基)哌啶-1-甲酸叔丁酯
[0686][0687]
在-78℃下向鏻盐(540mg,0.90mmol)在thf(5ml)中的溶液添加正丁基锂在thf中的溶液(2.5m,0.70ml,1.80mmol)。将反应混合物在40℃搅拌1小时。然后在0℃下向所述混合物添加化合物39(100mg,0.30mmol),然后升温至40℃2小时。将反应重复5次。将所述混合物用饱和nh4cl(80ml)淬灭并用etoac(100ml
×
3)萃取。将合并的有机层浓缩,并将残留物通过制备hplc进行纯化,得到作为白色固体的粗品化合物50(35mg,4%)。
[0688]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikma diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=2.104min;ms计算值:529,ms实测值:430[m h-boc]
。
[0689]
化合物51的合成:4-(2-((6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-17-氧络十六氢-1h-环戊烷并[a]菲-3-基)乙基)哌啶-1-甲酸叔丁酯
[0690][0691]
将化合物50(35mg,0.07mmol)、pd/c(10%,40mg)和pd(oh)2(20%,40mg)在etoac(2ml)中的混合物在h2下(在气球中)在室温搅拌过夜。将混合物过滤并将滤液浓缩,得到作为棕色固体的粗品化合物51(35mg,100%)。
[0692]
lcms柱:c18;柱尺寸:4.6
×
30mm 5μm;dikma diamonsil plus;流动相:b(acn):a(0.02%nh4ac 5%acn);梯度:3min内的(b%)-5-95-pos;流速1.5ml/min,停止时间3min。rt=2.126min;ms计算值:531,ms实测值:432[m h-boc]
。
[0693]
cvie412的合成:(6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-3-(2-(哌啶-4-基)乙基)十四氢-1h-环戊烷并[a]菲-17(2h)-酮
[0694][0695]
将化合物51(35mg,0.07mmol)在tfa/dcm(1:2,2ml)中的溶液在室温搅拌30分钟。将所述混合物用饱和nahco3稀释,以调整到ph 8-9。将混合物用dcm(25ml
×
3)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,产生作为黄色固体的化合物cvie412(13mg,46%)。
[0696]
cvie412的光谱数据:
[0697]1h nmr(cd3od,400mhz):δ4.04(s,1h),3.72-3.61(m,2h),3.09-3.06(m,2h),2.63(t,j=11.6hz,2h),2.48-2.41(m,1h),2.13-2.03(m,2h),1.75-1.66(m,5h),1.63-1.55(m,5h),1.45-1.35(m,3h),1.31-1.17(m,10h),1.10-1.03(m,2h),0.89(s,1h),0.87(s,3h),0.84(s,3h)。
[0698]
lcms柱:柱:c18;柱尺寸:4.6
×
50mm;流动相:b(acn):a(0.02%nh4ac);梯度:6.5min内的(b%)-5-95-pos;rt=3.203min;ms计算值:431,ms实测值:432[m h]
。
[0699]
化合物52的合成:((e)-5-((6s,10r,13s)-6-(羟甲基)-10,13-二甲基-7,17-二氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)戊基)(甲基)氨基甲酸叔丁酯
[0700][0701]
在-78℃下,向n-boc-n-甲基-5-三苯基鏻戊烯胺碘化物(4.26g,7.23mmol)在thf(50ml)中的混合物逐滴添加正丁基锂(3.18ml,7.95mmol)。将混合物在0℃搅拌20min。然后将混合物冷却至-30℃。然后向所述反应混合物添加化合物37(800mg,2.41mmol)。将所述混合物在室温搅拌过夜。将反应混合物用h2o淬灭并浓缩。将残留物在硅胶上通过柱层析进行纯化(pe/etoac=1/2),然后通过制备hplc进行纯化,产生作为无色油状物的化合物52(36mg,200mg)。
[0702]
化合物52的光谱数据:
[0703]1h nmr(cd3od,400mhz):5.16-5.12(m,1h),3.89-3.85(m,1h),3.77-3.73(m,1h),3.22-3.19(m,2h),2.83(s,3h),2.75-2.60(m,2h),2.60-2.50(m,2h),2.46-2.41(m,1h),2.33-2.27(m,1h),2.15-2.02(m,4h),1.90-1.79(m,2h),1.77-1.71(m,3h),1.60-1.52(m,4h),1.50(s,9h),1.45-1.42(m,1h),1.34-1.29(m,2h),1.26(s,3h),1.24-1.21(m,1h),1.19-1.05(m,2h),1.05-0.99(m,1h)。
[0704]
cvie401的合成:(6s,10r,13s,e)-6-(羟甲基)-10,13-二甲基-3-(5-(甲基氨基)亚戊基)十二氢-1h-环戊烷并[a]菲-7,17(2h,8h)-二酮
[0705][0706]
将化合物52(60mg,0.116mmol)在tfa/dcm(1ml/2ml)中的混合物在室温搅拌过夜。然后将混合物浓缩并用etoac稀释,用饱和na2co3洗涤,在na2so4上干燥,过滤并浓缩,产生作为黄色油状物的化合物cvie401(38mg,79%)。
[0707]
cvie401的光谱数据:
[0708]1h nmr(cd3od,400mhz):5.36(s,1h),3.89-3.86(m,1h),3.71-3.67(m,1h),2.80-2.77(m,2h),2.73-2.67(m,1h),2.55-2.39(m,6h),2.15-2.01(m,4h),1.92-1.85(m,1h),1.77-1.69(m,4h),1.65-1.45(m,7h),1.38-1.25(m,6h),1.19(s,3h),0.89(s,4h)。
[0709]
lcms:rt=2.128min,[m 1]
=416。
[0710]
化合物53的合成:(5-((6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-17-氧络十二氢-1h-环戊烷并[a]菲-3(2h,4h,10h)-亚基)戊基)(甲基)氨基甲酸叔丁酯
[0711][0712]
在-78℃下,向n-boc-n-甲基-5-三苯基鏻戊烯胺碘化物(4.39g,7.45mmol)在thf(45ml)中的混合物逐滴添加正丁基锂在thf中的溶液(4.46ml,2.5n,11.16mmol)。然后将混合物在0℃搅拌20min。将混合物冷却至-50℃并添加化合物39(830mg,2.48mmol)。将混合物在室温搅拌过夜。将所述混合物用h2o淬灭,浓缩,并通过柱层析进行纯化(pe/etoac=1/1),然后通过制备hplc进行纯化,得到作为白色固体的化合物53(80mg,300mg)。
[0713]
化合物53的光谱数据:
[0714]1h nmr(cd3od,400mhz):5.10-5.08(m,1h),4.05(s,1h),3.78-3.72(m,1h),3.23-3.20(m,2h),2.83(s,3h),2.56-2.53(m,1h),2.48-2.41(m,1h),2.12-2.10(m,1h),2.07-2.00(m,5h),1.85-1.81(m,1h),1.74-1.51(m,10h),1.49(s,9h),1.39-1.30(m,4h),1.21-1.18(m,1h),1.08-1.04(m,1h),0.96(s,3h),0.88(s,3h)。
[0715]
cvie406的合成:(6s,7s,10r,13s)-7-羟基-6-(羟甲基)-10,13-二甲基-3-(5-(甲基氨基)亚戊基)十四氢-1h-环戊烷并[a]菲-17(2h)-酮
[0716][0717]
将化合物53(80mg,0.155mmol)在tfa/dcm(1ml/2ml)中的溶液在室温搅拌10分钟。将所述混合物用饱和nahco3碱化至ph=8-9。将混合物用dcm(25ml
×
2)萃取。将合并的有机层在na2so4上干燥,过滤并浓缩。将残留物通过制备hplc进行纯化,得到作为黄色固体的化合物cvie406(60mg,94%)。
[0718]
lcms柱:rt=0.370min;ms计算值:417,ms实测值:418[m h]
。
[0719]
实施例2.用于测量生物活性的通用程序
[0720]
实施例动物护理
[0721]
本研究符合美国国立卫生研究院出版的《实验室动物的护理和使用指南》(guide of the care and use of laboratory animals)(nih第85-23号出版物,1996年修订)以及参与机构认可的动物护理指南。
[0722]
分离的左心室心肌细胞中的测量
[0723]
在使用酶溶液通过逆行冠状动脉灌注(rocchetti m等,j pharmacol exper therap 2005,313(1):207
–
215)分别从大鼠和豚鼠心室新鲜解离的肌细胞中,表征了化合物对(i)sr ca
2
摄入功能和(ii)动作电位(ap)的影响。
[0724]
统计分析
[0725]
完整动物实验:数据被报告为平均值
±
sd。统计分析通过学生t-检验(配对t检验)来进行。
[0726]
分离的肌细胞实验:数据被报告为平均值
±
se。对于重复测量值来说,通过双向anova对包括多个平均值的曲线进行比较;药物诱导的总体曲线陡度的变化根据“x因子组”相互作用的显著性来定义。由于ca
2
衰减的单指数拟合不充分,因此在报告了cat数据的少数几种细胞中未估算τ
decay
;样本量(n)被报告在相应的图中。stv对平均apd的依赖性通过线性回归来量化。在所有比较中,p《0.05被认为是统计显著的。
[0727]
实施例3.式(i)的化合物的体外筛选
[0728]
狗肾na
/k
atp酶活性的抑制
[0729]
正如本文中别处提到的,本发明的化合物是纯或显著纯serca2a刺激剂。因此,这些化合物对na
/k
atp酶的酶活性表现出很少或没有抑制。因此,测试了所述化合物对犬肾na
/k
atp酶的抑制效应。
[0730]
肾na
/k
atp酶的纯化按照描述的方法来进行(methods enzymol.1988,156:29-43)。在戊巴比妥麻醉(来自于意大利卫生部的进口授权0009171-09/04/2015-dgsaf-cod_uo-p,2015)下,从1-3岁的雄性比格犬(wuxi apptec,suzhou co.,ltd.1318wuzhong ave.,wuzhong district suzhou,215104p.r.china)切下肾脏。将肾脏切片,将外髓质切下并悬浮(1g/10ml)在含有250mm蔗糖、30mm组氨酸和5mm edta的ph 7.2的蔗糖-组氨酸溶液中。使用ultra turrax匀浆器将所述组织匀浆。将样品以6,000g离心15min。接下来,倒出上清液并以48,000g离心30min。将沉淀悬浮在蔗糖-组氨酸缓冲液中,并与溶解在含有25mm咪唑和1mm edta的ph 7.5的梯度缓冲液中的十二烷基硫酸钠(sds)溶液温育。将所述样品在蔗糖不连续梯度(10、15和29.4%)顶部分层,并以60,000g离心115min。将沉淀物悬浮在梯度缓冲液中。
[0731]
如(ferrandi m.等,hypertension 1996,28:1018-25)所述,通过测量
32
p从
32
p-atp的释放来测定na
/k
atp酶活性。将浓度逐渐提高的标准品哇巴因或测试化合物与0.3μg纯化的狗肾酶在终体积为120μl的培养基中,在37℃温育10min,所述培养基含有140mm nacl,3mm mgcl2,50mm hepes-tris,3mm atp,ph 7.5。然后添加10μl含有10mm kcl和20nci的
32
p-atp(3-10ci/mmol,perkin elmer)的溶液。允许反应在37℃继续15min,然后通过用20%v/v冰冷的高氯酸酸化来终止。使用活性炭(norit a,serva)通过离心分离
32
p,并对放射活性进行计数。抑制活性被表示为在不存在哇巴因或测试化合物的情况下进行的对照样品的百分数。使用多参数非线性回归最佳拟合程序(kaleidagraphtm,sinergy software)来计算引起na
/k
atp酶活性的50%抑制的化合物浓度(ic
50
)。
[0732]
如表1中所示,化合物cvie201、cvie202、cvie203、cvie204、cvie213、cvie214、cvie215、cvie216、cvie217、cvie218和cvie219不抑制纯化的na
/k
atp酶的酶活性,并且得到的以μm为单位表示的ic
50
》100μm。化合物cvie205、cvie206、cvie207、cvie208、cvie209、cvie210、cvie211、cvie212、cvie401、cvie402、cvie403、cvie404、cvie405、cvie406、cvie407、cvie408、cvie409、cvie410、cvie411和cvie412仅仅轻微抑制na
/k
atp酶(ic
50
范围在0.8至24μm之间)(表1)。
[0733]
已将化合物与参比药物地高辛(ic
50 0.18μm)和istaroxime(ic
50
0.14μm)进行比较(表1)。
[0734]
表1.狗肾na
/k
atp酶的抑制
[0735][0736][0737]
来自于正常豚鼠的心脏来源的sr微粒体中的serca2a atp酶活性
[0738]
也在源自于正常豚鼠心脏组织的sr微粒体中测试了浓度范围为1-200nm的本文公开的化合物刺激serca2a活性的能力。使用2月龄豚鼠(350-450g,来自于envigo,udine,italy)制备心脏serca2a微粒体。将豚鼠在戊巴比妥麻醉下处死。快速切下左心室(lv)并立即在液氮中冷冻。按照nediani c.等(j biol chem.1996,271:19066-7)描述的方法处理lv组织。将组织悬浮在4x体积的含有10mm nahco3、ph 7、1mm pmsf、10μg/ml抑肽酶和亮抑酶肽的缓冲液中,然后使用ultra turrax匀浆器匀浆。将样品以12,000g离心15分钟。将得到的上清液过滤,并以100,000g离心30min。通过将沉淀物用0.6m kcl、30mm组氨酸、ph 7悬浮
并进一步以100,000g离心30min,提取收缩性蛋白。将最终的沉淀物用0.3m蔗糖、30mm组氨酸、ph 7重构,并分成等分试样储存在-80℃下直至使用。
[0739]
正如(micheletti r.等,am j card 2007,99:24a-32a)所述,serca2a活性在体外被测量为在不存在和存在测试化合物的情况下,在不同ca
2
浓度(100-4000nm)下的
32
p-atp水解。将浓度逐渐提高的每种化合物(范围为1至200nm)与2μg富含serca2a的微粒体在80μl含有100mm kcl、5mm mgcl2、1μm a23187、20mm tris、ph 7.5的溶液中,在4℃预温育5min。然后添加20μl含有50nci
32
p-atp(3-10ci/mmol,perkin elmer)的5mm tris-atp。atp水解在37℃下继续15min,并通过使用100μl 20%v/v冰冷的高氯酸酸化来终止反应。使用活性炭(norit a,serva)通过离心分离
32
p,并测量放射活性。serca2a依赖性活性被鉴定为总水解活性被10μm环匹阿尼酸抑制的部分(seidler nw等,j biol chem.1989,264:17816-23)。
[0740]
通过使用s形方程拟合剂量-响应曲线,并计算最大速度下的活性(vmax)和ca
2
的kd(synergy software kaleidagraph 3.6)。化合物在正常豚鼠制备物中的效应被表示为在不存在化合物的情况下运行的对照样品的kd ca
2
的降低百分数(暗示对ca
2
亲和力的增加)(表2)。这种效应表明化合物在ca
2
浓度的生理范围内提高serca2a活性((rocchetti m等,j pharmacol exp ther.2005,313:207-15;rocchetti m等,j pharmacol exp ther.2008,326:957-65;ferrandi m等,br j pharmacol 2013,169:1849-1861)。数据为平均值
±
sd,n=实验次数,*至少p《0.05。
[0741]
在纳摩尔浓度下,测试化合物在来自于豚鼠心脏制备物的微粒体中降低ca
2
剂量响应曲线的serca2a kd ca
2
(表2)。这些结果表明所述化合物在ca
2
浓度的生理范围内提高serca2a活性,并暗示了松弛性效果。istaroxime被用作对比物,表明了它刺激serca2a的能力(表2)。与此不同,地高辛不能刺激serca2a活性(ferrandi m等,br j pharmacol 2013,169:1849-61;rocchetti m等,j pharmacol exp ther 2005,313:207-215)。
[0742]
表2.在来自于正常豚鼠的心脏来源的sr微粒体中测试化合物对serca2a kd ca
2
的影响
[0743]
[0744]
[0745][0746]
实施例4.在分离的心室肌细胞中对cvie214和cvie216的研究
[0747]
大鼠心室肌细胞中的sr ca
2
摄入功能
[0748]
为了测试化合物在舒张功能障碍模型中的效应,通过单次尾静脉注射链脲佐菌素(stz 50mg/kg,sigma-aldrich)使sprague dawley雄性大鼠(150-175g)变为糖尿病。stz被新鲜制备在ph 4.5的0.1m柠檬酸钠缓冲液中。在1周后测量空腹血糖,并且具有》300mg/dl的值的大鼠被认为患有糖尿病。在stz注射后9周,在分离的左心室肌细胞中评估药物对sr ca
2
摄入功能的影响。将肌细胞在特定药物存在下温育至少30min,以保证其细胞膜通透。统计分析通过“组比较”模型来进行。
[0749]
药物对sr ca
2
摄入速率的影响使用sr“载入方案”来评估,所述方案被特别设计用于排除na/ca交换剂(ncx)的贡献并评估从低水平的sr ca
2
载量开始的摄入速率。在电压钳条件下,通过落射荧光动态测量细胞内ca
2
浓度(fluo4-am)。同时记录膜电流,所述膜电流的时间依赖性成分主要反映i
cal
。所述sr载入方案包括通过短暂的咖啡因脉冲清空sr,然后通过激活ca
2
经肌膜ca
2
通道的内流(i
cal
)的电压步骤逐步重新填充sr。通过在细胞内和细胞外溶液中省略na
来阻断ncx。所述程序与已发表的方法相符,但稍有修改(rocchetti m等,j pharmacol exper therap 2005,313:207
–
215)。
[0750]
药物对sr ca
2
摄入的影响通过考察多种参数来分析:在所述载入方案期间1)ca
2
瞬时(cat)幅度和2)ca
2
诱导的ca
2
释放(cicr)增益增加的速率,其反映出sr重新填充和系统增益的速率,3)每次脉冲中胞质ca
2
衰减的时间常数(τ
decay
),反映出跨sr膜的净ca
2
转运速率(被serca2a)(τ
decay
的降低对应于sr ca
2
摄入增强)。
[0751]“载入方案”在检测serca2a激活中的特异性得到下述观察的支持,即它没有检测到地高辛的任何效应,所述地高辛是阻断na
/k
atp酶泵但没有serca2a刺激效应的变力性
药剂(rocchetti m等,j pharmacol exp ther 2005,313:207-215;alemanni m等,jmcc 2011,50:910-918)。
[0752]
cvie216(1μm)在sr重新装载期间提高ca
2
瞬时(cat)增加的速率(图1a);这与cicr增益的增加(图1b)和τ
decay
的降低(图1c)相关。
[0753]
cvie214(1μm)在sr载入方案中以与cvie216相似的方式改变cat参数(图2)。正如从serca2a增强所预期的,cicr增益(图2b)和τ
decay
(图2c)受到所述药物影响。cvie214在sr重新装载期间未能显著提高cat增加的速率(图2a)。然而,cicr增益的增加表明这可能反映出同时的i
cal
抑制而不是否定对serca2a的影响。
[0754]
图1和2中的结果合在一起表明cvie216和cvie214显著提高ca
2
被sr的摄入。在所使用的实验条件下,sr ca
2
摄入完全由serca2a支持;因此,所述结果与serca2a被所述两种药剂激活相一致。
[0755]
动作电位测量
[0756]
在豚鼠肌细胞中调节serca2a的1μm浓度下,评估了cvie216和cvie214对动作电位参数的影响。动作电位(ap)轮廓线提供了对膜离子通道的综合功能的第一线评估,并且其变化可能揭示了附带作用——可能导致化合物的不良效应。为了提高ap轮廓线作为报告物的灵敏度,还测试了对ap参数的刺激率依赖性的影响,从而提供了一种多参数(更严格)的方法。ap在豚鼠心室肌细胞中记录,因为所述ap轮廓可重现人类的ap。将肌细胞在在药物存在下温育至少30min,以确保即使长时间暴露也没有影响。统计分析通过“组比较”模型来进行。
[0757]
豚鼠心室肌细胞中的ap在正常的tyrode溶液中,在36.5℃下记录。在4个刺激率(0.5-1-2-4hz)下测量了下述参数:舒张期膜电位(e
diast
),最大去极化速度(dv/dt
max
),动作电位持续时间(90%复极化时的apd)。稳态起搏期间的短期apd变动(stv)这个复极化稳定性的指标,被测量为与apdnfapd
n 1
图(poincare图)中的恒等线的绝对正交偏差之和(altomare c等,circulation a&e 2015,8:1265-1275)。
[0758]
在1μm下,cvie216(图3a)和cvie214(图4a)不影响动作电位持续时间(apd
90
)、舒张期膜电位(e
diast
)、最大去极化速度(dv/dt
max
)和每种ap参数的刺激率依赖性。
[0759]
短期apd变动(stv)是电不稳定性的标志物,并与致心律失常风险相关。stv是平均apd的函数;因此,stv在多个起搏频率(0.5-1-2和4hz)下测量,以将其评估扩展到较宽的apd范围。stv及其对平均apd的依赖性不受cvie216(图3b)和cvie214(图4b)两者的显著影响。
[0760]
总而言之,用于动作电位分析的多参数方法表明不存在对心脏电活动的不良药物影响。因此,根据该分析,cvie216和cvie214选择性地执行serca2a调节(正性松弛性药物),即不影响电活动和所涉及的膜电流。
[0761]
实施例5.对cvie214和cvie216的体内研究
[0762]
大鼠中的生物可利用性
[0763]
大鼠中的生物可利用性由中国的sundia meditech service测量。具体来说,在以1mg/kg的剂量静脉内注射(i.v.)和以10mg/kg的剂量口服给药(os)后测量cvie214-盐和cvie216-盐在大鼠中的生物可利用性。测试化合物化合物cvie214-盐和cvie216-盐的血浆浓度从0时至24h时以一定间隔测量,并通过lc-ms方法检测。计算f值(%),对于cvie214-盐
和cvie216-盐来说结果分别为41.5%和16.9%。
[0764]
小鼠中的急性毒性
[0765]
在小鼠(albino swiss cd-1,体重30g)中确定了测试化合物cvie214-盐和cvie216-盐的急性毒性。将化合物以逐渐增加的剂量口服给药或静脉内注射,以鉴定导致50%死亡的剂量。死亡发生在给药后30min内,存活则在24h后。
[0766]
cvie214-盐和cvie216-盐急性毒性的结果报告在表3中。作为比较,也包括了参比化合物地高辛和istaroxime的急性毒性。地高辛参考文献数据(www.lookchem.com,“静脉内地高辛参考手册”(reference for digoxin intravenous):afifi am,ammar em.pharmacological research communications.vol.6,pg.417,1974;“口服地高辛参考手册”(reference for digoxin oral):archives internationales de pharmacodynamie et de therapie.第153卷,第436页,1965)(表3)。
[0767]
表3.cvie214-盐和cvie216-盐在小鼠中的急性毒性(ld
50
)
[0768][0769]
在链脲佐菌素糖尿病大鼠中的血液动力学(超声心动图2m-多普勒-组织多普勒)
[0770]
在糖尿病大鼠模型中测试了cvie214和cvie216。简单来说,将大鼠用链脲佐菌素(stz)注射。从stz注射起7-9周后,使大鼠经受在尿烷麻醉下进行的经胸超声心动图和多普勒评估。根据美国超声心动图学会(american society of echocardiography)指南(lang rm等,eur j echocardiography 2006,7:79-108),使用二维引导的m模式记录来获得左心室舒张末期直径(lvedd)、左心室收缩末期直径(lvesd)、舒张期后部(pw)和间隔(sw)壁厚度的短轴测量值。缩短分数被计算为fs=(lvedd-lvesd)/lvedd。相对壁厚被计算为pwtd ivstd/lvedd。从心尖四腔视图通过脉冲多普勒在二尖瓣小叶的顶端处测量二尖瓣流入,以获得早期和晚期充盈速度(e、a)以及早期充盈速度的减速时间(dt)。减速斜率被计算为e/dt比率。二尖瓣减速指数被计算为dt/e比率。从心尖四腔视图评估组织多普勒成像(tdi),以记录间隔二尖瓣环形运动、即心肌收缩峰值(s’)和舒张早期和晚期速度(e’和a’)。
[0771]
在获取基线血液动力学测量值后,向大鼠给药地高辛、cvie214、cvie216,并与对照进行比较。将用作参比药物的地高辛以0.11mg/kg/min的速率静脉内输注15min,并在1h
后测量超声心动图参数。将cvie214-盐和cvie216-盐以0.2mg/kg/min的速率静脉内输注到stz糖尿病大鼠中,并在15min和30min后测量超声心动图参数。
[0772]
表4-6示出了地高辛、cvie214-盐和cvie216-盐在stz糖尿病大鼠中的血液动力学参数。表4-6中示出的数据是平均值
±
sd;带有星号的值是统计显著的,具有至少p《0,05。
[0773]
所述数据表明,所述链脲佐菌素糖尿病大鼠模型与健康的对照大鼠相比以舒张功能障碍为特征(对照n=18只大鼠;stz n=20只大鼠)(表4)。具体来说,stz大鼠显示出dt、dt/e的增加和e、dt/e、e’、hr的降低。cvie214-盐和cvie216-盐改善在stz中与对照相比恶化的舒张功能(表4),引起dt、dt/e的显著降低以及e/dt和e’的增加,并伴有sv和co改善(表5-6)。从cvie216-盐输注起30min后e/e’显著降低(表6)。15min后只有cvie214轻度但显著地提高s’和hr(表5)。作为参比化合物的地高辛改善舒张功能,降低dt、dt/e并提高e/dt、e’和收缩功能(fs、s’),但不影响总体心功能例如sv和co(表4)。
[0774]
表4.对照和stz糖尿病大鼠中的血液动力学参数和地高辛iv输注在stz大鼠中的效应
[0775][0776][0777]
fs%:缩短分数,收缩功能;e m/s:二尖瓣流入的早期充盈速度;a m/s:二尖瓣流入的晚期充盈速度;e/a:lv功能的指数;dt ms:e波减速时间;dt/e s2/m:二尖瓣减速指数;e/dt m/s2:减速斜率;s'cm/s tdi:收缩速度;e'cm/s tdi:早期松弛速度;a'cm/s tdi:晚期松弛速度;e/e’:lv充盈压的指数;co ml/min:心输出量;hr搏/min:心率;sv ml/搏:每搏输出量。*至少p《0.05,对照相比于stz或stz加上药物相比于stz之前。
[0778]
表5.stz糖尿病大鼠在cvie214-盐iv输注后的血液动力学参数
[0779][0780]
表6.stz糖尿病大鼠在cvie216-盐iv输注后的血液动力学参数
[0781][0782][0783]
报告物结合测定
[0784]
放射性配体与一组受体的结合,在粗膜制备物上通过eurofins,按照已发表的程序并使用适合的参比标准品(eurofin,taiwan,化合物编码cvie216-3(1226840),研究编号tw04-0004235,查询编号tw04-0004235-q04,cvie therapeutics limited,taiwan)来进行。cvie216-盐以10μm的浓度进行测试。正如表7中所示,在一组受体上没有记录到显著的相互作用。
[0785]
表7cvie216-盐的受体结合测定
[0786]
[0787]
[0788]
[0789][0790]
注:bov=牛;ham=仓鼠;hum=人类;没有项目符合显著性》50%刺激或抑制的判定标准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
