1.本发明涉及蒽环类衍生物及其在与靶结合分子(包括但不限于抗体)形成结合物中的用途。还提供了靶结合分子和蒽环类衍生物的结合物。
背景技术:
2.小分子量毒素与特异性结合蛋白(如抗体)的结合物是将毒性有效载荷导向体内目标的有力工具。一个实例是使用这种结合物使毒性有效载荷靶向癌细胞,这种结合物在癌症治疗中显示出巨大的前景。
3.为了开发有效和安全的用于癌症治疗的结合物,需要解决几个方面。首先,结合蛋白或抗体需要对正常或健康组织细胞很难表达或理想情况下不应表达的给定的肿瘤特异性抗原(tsa)具有特异性。
4.第二,药物和结合蛋白之间的共价键或连接需要在循环中保持稳定,以防止毒性有效载荷在血流中意外释放,但它必须在结合和/或内化到癌细胞后有效释放药物。第三,毒性有效载荷必须具有足够高的毒性或效力,以实现对癌细胞的破坏,即使癌细胞上表达的tsa数量可能有限且因此只能内化有限数量的adc,或者在与癌细胞结合或内化到癌细胞时毒性有效载荷的释放没有以足够高的效率实现。
5.蒽环类衍生物pnu-159682被描述为奈莫柔比星的代谢物(quintieri等人,(2005)clin.cancer res.11,1608-1617),且据报道对一个卵巢(a2780)细胞系和一个乳腺癌(mcf7)细胞系在皮摩尔至飞摩尔范围内的体外细胞杀伤表现出极高的效力(wo2012/073217a1)。pnu-159682的衍生物也已在wo2016/102679中描述。
6.pnu-159682衍生物与抗体的结合物描述于wo2009/099741、wo2016/127081和wo2016/102679,yu等人,clin.cancer res 2015,21,3298和stefan等人,mol.cancer.ther.,2017,16,879。
技术实现要素:
7.本发明提供适用于药物结合物的蒽环类(pnu)衍生物。具体而言,提供了pnu159682的衍生物,其缺乏c14碳和附着的羟基官能团,其中乙二胺(eda)基团形成pnu159682的c13羰基和马来酰亚胺基团之间的连接区域的一部分。当连接基团包含val-cit-pab时,马来酰亚胺基团可被适合于共轭反应的任何反应性基团取代。这种有效载荷能够与另一分子上的游离硫醇基团反应。当游离硫醇位于蛋白质上时,可形成蛋白质-药物结合物(pdc)。
8.因此,在第一方面中提供一种式(i)的蒽环类(pnu)衍生物:
[0009][0010]
其中[x]是选自包括取代的或未被取代的烷基基团、取代的或未被取代的杂烷基基团、取代的或未被取代的芳基基团、取代的或未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基;
[0011]
[l1]和[l2]是选自由缬氨酸(val)、瓜氨酸(cit)、丙氨酸(ala)、天冬酰胺(asn)、肽、-(ch2)
n-、-(ch2ch2o)
n-、对氨基苄氧基羰基(pab)、val-cit-pab、val-ala-pab、ala-ala-asn-pab、除甘氨酸外的任何氨基酸及其组合组成的组的任选连接基团。
[0012]
式(i)的蒽环类(pnu)衍生物可包含[l1]、[l2]或[l1]和[l2]。
[0013]
优选地,[l1]和/或[l2]是肽时,所述肽不包含甘氨酸。
[0014]
本领域技术人员将清楚,当缺少可选间隔基和/或可选连接基团时,在它们的位置上保留键。
[0015]
优选地,[x]选自包括聚乙二醇、
[0016]
的组,其中表示与分子的其余部分连接的点并且其中[r]是选自包括取代的或未被取代的烷基基团、取代的或未被取代的杂烷基基团、取代的或未被取代的芳基基团、取代的或未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基。
[0017]
最优选地,[x]是聚乙二醇。聚乙二醇可以是peg4。
[0018]
优选地,[l2]是对氨基苄氧基羰基(pab)或丙氨酸。
[0019]
优选地,pnu衍生物具有选自以下结构的结构:
[0020][0021][0022]
在第二方面中,提供一种式(v)的蒽环类(pnu)衍生物:
[0023][0024]
其中[x]是选自包括取代的或未被取代的烷基基团、取代的或未被取代的杂烷基基团、取代的或未被取代的芳基基团、取代的或未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基;
[0025]
其中[z]是反应性基团。反应性基团可以是适于在共轭反应,特别是与靶结合分子的共轭反应中使用的任何反应性基团。
[0026]
[z]因此可以是包含用于生物共轭反应的官能团的部分。用于生物共轭反应的官能团包括但不限于,
[0027]
通过硫醚和硒醇醚反应与蛋白质上的硫醇基团或硒醇基团反应的马来酰亚胺或烷基卤化物;
[0028]
用于与马来酰亚胺、烷基卤化物或硫醇官能化分子(包括蛋白质半胱氨酸残基的硫醇基团)反应的巯基基团;
[0029]
用于通过硫醇-二硫化物交换与硫醇基团反应形成二硫键的活性二硫化物,如吡啶基二硫醇(npys硫醇)或tnb硫醇(5-硫醇-2-硝基苯甲酸);
[0030]
通过酰胺键形成反应连接到蛋白质和生物分子上的羧基基团的氨基基团;
[0031]
通过菌株促进的炔烃-叠氮化物环加成无铜化学与叠氮官能化生物分子反应的炔烃基团,特别是环约束炔烃,如二苯并环辛炔(dbco)或双环[6.1.0]壬炔(bcn)。叠氮官能团可通过例如将非天然氨基酸对叠氮基甲基-l-苯丙氨酸并入而引入蛋白质中,或使用酶介导的糖工程将叠氮官能团引入蛋白质聚糖中以连接含叠氮基的糖类似物;
[0032]
通过菌株促进的炔烃-叠氮化物环加成无铜化学与炔烃官能化靶结合分子反应的叠氮基基团;
[0033]
通过肟形成连接与生物分子上的醛基和酮基反应的氨基氧基基团。酮可以通过使用琥珀终止密码子技术引入蛋白质中,例如引入非天然氨基酸、对乙酰苯丙氨酸。醛可通过还原糖的存在而在生物分子上找到,并可通过n-末端丝氨酸残基的高碘酸盐氧化或碳水化合物顺式乙二醇基团的高碘酸盐氧化引入蛋白质。醛基还可以通过特定序列内的蛋白质半胱氨酸经甲酰甘氨酸生成酶转化为甲酰甘氨酸而并入蛋白质中。另外,含甲酰甘氨酸的蛋白质经由hydrazino-pictet-spengler(hips)连接与有效载荷结合;
[0034]
用于通过肟或肼键形成连接反应与氨基氧基或酰肼或肼基官能化生物分子反应的醛或酮基团。蛋白质氨基氧基和酰肼功能化的蛋白质可以通过内含肽-融合蛋白的裂解产生。
[0035]
[z]因此可选自由马来酰亚胺、烷基卤化物、巯基基团、活性二硫化物(如吡啶基二硫醇(npys硫醇)或tnb硫醇(5-硫醇-2-硝基苯甲酸))、氨基基团、炔基基团(如环约束炔烃,如二苯并环辛炔(dbco)或双环[6.1.0]壬炔(bcn))、叠氮基基团、氨氧基基团、醛基和酮基组成的组。
[0036]
[z]也可以是酶介导的生物共轭反应的一部分。用于酶介导的共轭反应的部分包括但不限于用于sortase酶介导的抗体结合的polygly[(gly)n]或用于细菌转谷氨酰胺酶介导的与含有序列(如lys-lys-gln-gly和lys-pro-glu-thr-gly)的谷氨酰胺-羧酰胺基团结合的适当伯胺。
[0037]
[z]因此可选自由polygly和伯胺组成的组。
[0038]
根据本发明第二方面的pnu衍生物因此可对应于本发明第一方面的pnu衍生物,其中l1是val-cit-pab,l2不存在,且其中马来酰亚胺基团可替换为上文定义的另一反应性基团。
[0039]
优选地,[x]选自包括聚乙二醇、优选地,[x]选自包括聚乙二醇、的组,其中表示与分子的其余部分连接的点并且其中[r]是选自包括取代的或未被取代的烷基基团、取代的或未被取代的杂烷基基团、取代的或未被取代的芳基基团、取代的或未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基。
[0040]
最优选地,[x]是聚乙二醇。聚乙二醇可以是peg4。
[0041]
第一方面的pnu衍生物可以与多种部分结合。具体而言,第一方面的pnu衍生物可与结合靶的分子(在本文中称为靶结合分子)结合。第二方面的pnu衍生物可与多种部分结合。特别是,第二方面的pnu衍生物可与结合靶的分子(在本文中称为靶结合分子)结合。靶结合分子的实例包括但不限于生物分子、肽、小分子、蛋白质和核酸(包括但不限于适体)。在一些情况下,靶结合分子可以是多聚体(例如二聚体、三聚体和高阶多聚体或多亚基蛋白质)。
[0042]
因此,在又一方面中,提供一种包含根据第一方面的pnu和结合分子的靶结合分子-药物结合物。作为选择,在该方面中提供一种包含根据第二方面的pnu和结合分子的靶结合分子-药物结合物。适合在该方面中使用的结合分子的实例包括但不限于生物分子、肽、小分子、蛋白质和核酸(包括但不限于适体)。根据该方面,提供一种靶结合分子-药物结合物,其包含靶结合分子和蒽环类(pnu)衍生物,其中靶结合分子-药物结合物具有式(ii)的结构:
[0043][0044]
其中[x]是选自包括取代的或未被取代的烷基基团、取代的或未被取代的杂烷基基团、取代的或未被取代的芳基基团、取代的或未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基;
[0045]
[l1]和[l2]是选自由缬氨酸(val)、瓜氨酸(cit)、丙氨酸(ala)、天冬酰胺(asn)、肽、-(ch2)
n-、-(ch2ch2o)
n-、对氨基苄氧基羰基(pab)、val-cit-pab、val-ala-pab、ala-ala-asn-pab、除甘氨酸外的任何氨基酸及其组合组成的组的任选连接基团;和
[0046]
y是靶结合分子。
[0047]
式(ii)的靶结合分子-药物结合物可包含[l1]、[l2]或[l1]和[l2]。
[0048]
优选地,靶结合分子-药物结合物,其中,[l1]和/或[l2]是肽,所述肽不包含甘氨酸。
[0049]
本领域技术人员将清楚,当缺少可选间隔基和/或可选连接基团时,在它们的位置上保留键。
[0050]
在一个实施方式中,蒽环类(pnu)衍生物包含[l1]和/或[l2],且[x]是任选的。因此,[l1]和/或[l2]可以是选自由缬氨酸(val)、瓜氨酸(cit)、丙氨酸(ala)、天冬酰胺(asn)、肽、-(ch2)
n-、-(ch2ch2o)
n-、对氨基苄氧基羰基(pab)、val-cit-pab、val-ala-pab、ala-ala-asn-pab、除甘氨酸外的任何氨基酸及其组合组成的组的连接基团。式(i)的蒽环类(pnu)衍生物可包含[l1]、[l2]或[l1]和[l2]。式(i)的蒽环类(pnu)衍生物可包含[l1]和/或[l2]。
[0051]
在一个实施方式中,提供一种式(i)的蒽环类(pnu)衍生物:
[0052][0053]
其中[x]是选自包括取代的或未被取代的烷基基团、取代的或未被取代的杂烷基基团、取代的或未被取代的芳基基团、取代的或未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基;
[0054]
[l1]和/或[l2]是选自由缬氨酸(val)、瓜氨酸(cit)、丙氨酸(ala)、天冬酰胺(asn)、肽、-(ch2)
n-、-(ch2ch2o)
n-、对氨基苄氧基羰基(pab)、val-cit-pab、val-ala-pab、ala-ala-asn-pab、除甘氨酸外的任何氨基酸及其组合组成的组的连接基团;
[0055]
其中式(i)的蒽环类(pnu)衍生物包含[l1]、[l2]或[l1]和[l2]。
[0056]
优选地,[x]选自包括聚乙二醇、
[0057]
的组,其中表示与分子的其余部分连接的点并且其中[r]是选自包括取代的或未被取代的烷基基团、取代的或未被取代的杂烷基基团、取代的或未被取代的芳基基团、取代的或未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基。
[0058]
最优选地,[x]是聚乙二醇。聚乙二醇可以是peg4。
[0059]
优选地,[l2]是对氨基苄氧基羰基(pab)或丙氨酸。
[0060]
优选地,pnu衍生物具有选自以下结构的结构:
[0061][0062][0063]
根据该方面,还提供一种靶结合分子-药物结合物,其包含靶结合分子和蒽环类(pnu)衍生物,其中靶结合分子-药物结合物具有式(v)的结构:
[0064][0065]
其种[x]是选自包括取代的或未被取代的烷基基团、取代的或未被取代的杂烷基基团、取代的或未被取代的芳基基团、取代的或未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基;
[0066]
[z]是源自用于使蒽环类(pnu)衍生物与靶结合分子结合的反应性基团的连接基团;和
[0067]
y是靶结合分子。
[0068]
[z]通常是源自用于使蒽环类(pnu)衍生物与靶结合分子结合的反应性基团的部分。[z]可以是源自选自由马来酰亚胺、烷基卤化物、巯基基团、活性二硫化物、氨基基团、炔基基团、叠氮基基团、氨氧基基团、醛基和酮基组成的组的反应性基团的部分。
[0069]
[z]因此可选自由二硫键、酰胺键、肟键、腙键、硫醚键、1,2,3三唑和polygly组成的组。
[0070]
优选地,[x]选自包括聚乙二醇、优选地,[x]选自包括聚乙二醇、的组,其中表示与分子的其余部分连接的点并且其中[r]是选自包括取代的或未被取代的烷基基团、取代的或未被取代的杂烷基基团、取代的或未被取代的芳基基团、取代的或未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基。
[0071]
最优选地,[x]是聚乙二醇。聚乙二醇可以是peg4。
[0072]
优选地,靶结合分子是蛋白质或核酸。靶结合蛋白(也可称为特异性抗原结合蛋白)的实例包括但不限于免疫球蛋白或抗体、免疫球蛋白fc区、免疫球蛋白fab区、fab’、fv、fv-fc、单链fv(scfv)、scfv-fc、(scfv)2、双特异抗体、三特异抗体、四特异抗体、双特异性t-细胞结合器(bite)、内含肽、vnar结构域、单结构域抗体(sdab)、vh结构域或支架蛋白质(亲和体、centyrin、darpin等)。靶结合核酸的实例包括但不限于适体。
[0073]
优选地,靶结合分子-药物结合物为蛋白质,蒽环类(pnu)衍生物与蛋白质氨基酸序列中含硫醇的氨基酸残基结合或与通过蛋白质化学修饰引入的硫醇基结合,例如,在特定抗原结合蛋白的氨基酸序列的n末端或c末端并入。硫醇基团也可以引入其他靶结合分子,如核酸。
[0074]
靶结合蛋白(也可称为特异性抗原结合蛋白)可选自包括免疫球蛋白或抗体、免疫球蛋白fc区、免疫球蛋白fab区、fab’、fv、fv-fc、单链fv(scfv)、scfv-fc、(scfv)2、双特异抗体、三特异抗体、四特异抗体、双特异性t-细胞结合器(bite)、内含肽、vnar结构域、单结构域抗体(sdab)、vh结构域或支架蛋白质(亲和体、centyrin、darpin等)的组。
[0075]
在优选的实施方式中,靶结合分子可包括特异性抗原结合蛋白,可包含由式(iii)表示的氨基酸序列:
[0076]
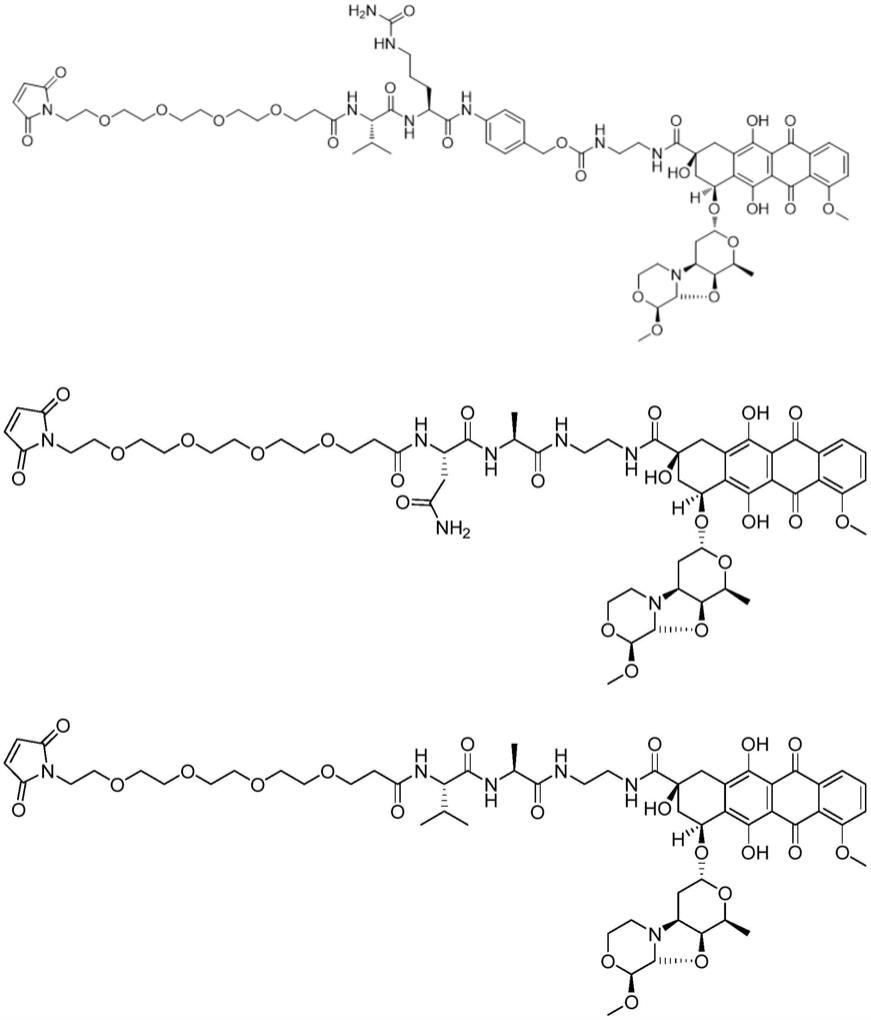
fw1-cdr1-fw2-hv2-fw3a-hv4-fw3b-cdr3-fw4
ꢀꢀ
(iii)
[0077]
其中
[0078]
fw1是框架区
[0079]
cdr1是cdr序列
[0080]
fw2是框架区
[0081]
hv2是高变序列
[0082]
fw3a是框架区
[0083]
hv4是高变序列
[0084]
fw3b是框架区
[0085]
cdr3是cdr序列
[0086]
fw4是框架区。
[0087]
优选地,特异性抗原结合蛋白与受体酪氨酸激酶样孤儿受体1(ror1)结合。优选地,ror1-特异性抗原结合分子不与受体酪氨酸激酶样孤儿受体2(ror2)结合。优选地,ror1-特异性抗原结合分子与人ror1和小鼠ror1(mror1)二者结合。优选地,ror1-特异性抗原结合分子与去糖基化ror1结合。此类分子在待审的国际专利申请号pct/ep2018/086823中描述,其内容通过引用并入本文。
[0088]
更优选地,ror1-特异性抗原结合分子不与选自以下序列的线性肽序列结合:
[0089]
ymeslhmqgeienqi(seq id no:34)
[0090]
cqpwnsqyphthtftalrfp(seq id no:35)
[0091]
rstiygsrlrirnldttdtgyfq(seq id no:36)
[0092]
qcvatngkevvsstgvlfvkfgppptaspgysdeye(seq id no:37)
[0093]
在靶结合分子-药物结合物的该实施方式中,特异性抗原结合蛋白可包括:
[0094]
fw1是20至28个氨基酸的框架区
[0095]
cdr1是选自dtsyglys(seq id no:1)、gakyglaa(seq id no:2)、gakyglfa(seq id no:3)、ganyglaa(seq id no:4)或ganyglas(seq id no:5)的cdr序列
[0096]
fw2是6至14个氨基酸的框架区
[0097]
hv2是选自ttdwermsig(seq id no:6)、ssnqerisis(seq id no:7)或ssnkeqisis(seq id no:8)的高变序列
[0098]
fw3a是6至10个氨基酸的框架区
[0099]
hv4是选自nkrak(seq id no:9)、nkrtm(seq id no:10)、nkgak(seq id no:11)或nkgtk(seq id no:12)的高变序列
[0100]
fw3b是17至24个氨基酸的框架区
[0101]
cdr3是选自qsgmaistgsghgynwy(seq id no:13)、qsgmaidigsghgynwy(seq id no:14)、ypwamwgqwy(seq id no:15)、vfmpqhwhpaahwy(seq id no:16)、rearhpwlrqwy(seq id no:17)或ypwgagapwlvqwy(seq id no:18)的cdr序列
[0102]
fw4是7至14个氨基酸的框架区
[0103]
或其具有至少45%序列同一性的功能变体,
[0104]
更优选地,fw1选自:
[0105]
asvnqtprtatketgesltincvlt(seq id no:19),
[0106]
akvdqtprtatketgesltincvlt(seq id no:20),
[0107]
trvdqtprtatketgesltincvvt(seq id no:21),
[0108]
trvdqtprtatketgesltincvlt(seq id no:22),
[0109]
asvnqtprtatketgesltincvvt(seq id no:23),
[0110]
trvdqspsslsasvgdrvtitcvlt(seq id no:24)或asvtqsprsasketgesltitcrvt(seq id no:56),fw2选自:tswfrknpg(seq id no:25)或tywyrknpg(seq id no:26);fw3a选自:gryvesv(seq id no:27)或grysesv(seq id no:28),fw3b选自:sfslrikdltvadsatyycka(seq id no:29)、sftltisslqpedsatyycra(seq id no:30)、sftltisslqpedfatyycka(seq id no:31)或sfslrissltvedsatyycka(seq id no:57),以及fw4选自:dgagtvltvn(seq id no:32)、dgagtkveik(seq id no:33)或dgqgtklevk(seq id no:58);或其具有至少45%序列同一性的功能变体。
[0111]
更优选地,ror1-特异性抗原结合分子包含选自以下序列的氨基酸序列:
[0112]
asvnqtprtatketgesltincvltdtsyglystswfrknpgttdwermsiggryvesvnkraksfslrikdltvadsatyyckaqsgmaistgsghgynwydgagtvltvn(seq id no:39);
[0113]
akvdqtprtatketgesltincvltdtsyglystswfrknpgttdwermsiggryvesvnkraksfslrikdltvadsatyyckaqsgmaidigsghgynwydgagtvltvn(seq id no:40);
[0114]
trvdqtprtatketgesltincvvtgakyglaatywyrknpgssnqerisisgryvesvnkrtmsfslrikdltvadsatyyckaypwamwgqwydgagtvltvn(seq id no:41);
[0115]
trvdqtprtatketgesltincvvtgakyglfatywyrknpgssnqerisisgryvesvnkrtmsfslrikdltvadsatyyckavfmpqhwhpaahwydgagtvltvn(seq id no:42);
[0116]
trvdqtprtatketgesltincvltdtsyglystswfrknpgttdwermsiggryvesvnkgaksfslrikdltvadsatyyckarearhpwlrqwydgagtvltvn(seq id no:43);
[0117]
asvnqtprtatketgesltincvvtganyglaatywyrknpgssnqerisisgryvesvnkrtmsfslrikdltvadsatyyckaypwgagapwlvqwydgagtvltvn(seq id no:44);
[0118]
trvdqspsslsasvgdrvtitcvltganyglastywyrknpgssnkeqisisgrysesvnkgtksftltisslqpedsatyycraypwgagapwlvqwydgagtkveik(seq id no:45);
[0119]
trvdqspsslsasvgdrvtitcvltganyglastywyrknpgssnqerisisgrysesvnkrtmsftltisslqpedsatyycraypwgagapwlvqwydgagtkveik(seq id no:46);
[0120]
trvdqspsslsasvgdrvtitcvltdtsyglystswfrknpgttdwermsiggryvesvnkgaksftl
tisslqpedfatyyckarearhpwlrqwydgagtkveik(seq id no:47);
[0121]
trvdqspsslsasvgdrvtitcvltdtsyglystywyrknpgssnkeqisisgrysesvnkgtksftltisslqpedsatyycrarearhpwlrqwydgagtkveik(seq id no:48);
[0122]
trvdqspsslsasvgdrvtitcvltdtsyglystywyrknpgttdwermsiggrysesvnkgaksftltisslqpedsatyycrarearhpwlrqwydgagtkveik(seq id no:49);
[0123]
asvtqsprsasketgesltitcrvtganyglaatywyrknpgssnqerisisgrysesvnkrtmsfslrissltvedsatyyckaypwgagapwlvqwydgqgtklevk(seq id no:59);
[0124]
或其具有至少45%序列同一性的功能变体。
[0125]
ror1-特异性抗原结合分子可以被人源化。ror1-特异性抗原结合分子可以被去免疫。
[0126]
ror1-特异性抗原结合分子还可以是融合蛋白的一部分。优选的融合蛋白是融合到免疫球蛋白fc区的ror1特异性抗原结合分子。优选地,免疫球蛋白fc区是人免疫球蛋白fc区。在一些情况下,ror1-特异性抗原结合分子可以是二聚体、三聚体和高阶多聚体。这种多聚体也可以是与其他分子的融合蛋白,包括但不限于免疫球蛋白fc。融合蛋白的各个结构域可以通过可选的连接基团连接。连接基团可包括但不限于(g4s)5、pgvqpspggggs(称为wbg4s)(seq id no:50)和pgvqpapggggs(称为wbg4sgm)(seq id no:51)。
[0127]
本文还提供了根据上述方面的用于治疗的靶结合分子-药物结合物。
[0128]
本文还提供了根据上述方面的用于治疗癌症的靶结合分子-药物结合物。
[0129]
本文还提供了根据上述方面的靶结合分子-药物结合物在制造用于治疗有需要的患者的疾病的药物中的用途。
[0130]
本文还提供了一种治疗需要治疗的患者的疾病的方法,所述方法包括对所述患者施用治疗有效剂量的根据上述方面的靶结合分子-药物结合物。所述疾病可以是癌症。
[0131]
优选地,癌症是ror1-阳性的癌症类型。更优选地,癌症选自包括血癌如淋巴瘤和白血病、慢性淋巴细胞白血病(cll)、套细胞淋巴瘤(mcl)、b细胞急性淋巴细胞白血病(b-all)、边缘区淋巴瘤(mzl)、非霍奇金淋巴瘤(nhl)、急性髓细胞白血病(aml)和实体瘤包括神经母细胞瘤、肾癌、肺癌、结肠癌、卵巢癌、胰腺癌、乳腺癌、皮肤癌、子宫癌、前列腺癌、甲状腺癌、头颈癌、膀胱癌、胃癌或肝癌的组。癌症可以是间皮瘤或三阴性乳腺癌(tnbc)。间皮瘤可能是胸膜间皮瘤。
[0132]
在上述方面的实施方式中,靶结合分子是抗体。在上述方面的另一个实施方式中,靶结合分子结合her-2。优选地,靶结合分子是结合her-2的抗体。更优选地,抗体是曲妥珠单抗或其衍生物。
[0133]
本文还提供了一种药物组合物,其包含根据上述任一方面的靶结合分子-药物结合物和至少一种其他药学上可接受的成分。
附图说明
[0134]
图1-由val-cit(vc)pab pnu结合物释放的有效载荷的实例。
[0135]
图2-本发明的pnu衍生物。
[0136]
图3-与非结合对照(2vhfc-na-eda-pnu)相比b1hfc-na-eda-pnu结合物对ror1阳性pa-1细胞系的杀伤效力。
[0137]
图4-与非结合对照(2vhfc-va-eda-pnu)相比b1hfc-va-eda-pnu结合物对ror1阳性pa-1细胞系的杀伤效力。
[0138]
图5-与非结合对照(2vhfc-va-eda-pnu)相比b1hfc-na-eda-pnu结合物对ror1敲除的pa-1细胞系的杀伤效力。敲除ror1的pa-1细胞系中的b1hfc-na-eda-pnu结合物未显示出细胞杀伤。
[0139]
图6-与非结合对照(2vhfc-va-eda-pnu)相比b1hfc-va-eda-pnu结合物对敲除ror1的pa-1细胞系的杀伤效力。敲除ror1的pa-1细胞系中的b1hfc-va-eda-pnu结合物未显示出细胞杀伤。
[0140]
图7-与非结合对照(2vhfc-na-eda-pnu)相比,b1hfc-na-eda-pnu结合物对kasumi-2细胞系的杀伤效力。
[0141]
图8-与非结合对照(2vhfc-va-eda-pnu)相比b1hfc-va-eda-pnu结合物对kasumi-2细胞系的杀伤效力。
[0142]
图9-与非结合对照(2vhfc-na-eda-pnu)相比b1hfc-na-eda-pnu结合物对mhh-es1细胞系的杀伤效力。
[0143]
图10-与非结合对照(2vhfc-va-eda-pnu)相比b1hfc-va-eda-pnu结合物对mhh-es1细胞系的杀伤效力。
[0144]
图11-与非结合对照(2vhfc-vc-pab-eda-pnu)相比b1hfc-vc-pab-eda-pnu结合物对ror1阳性pa-1细胞系的杀伤效力。
[0145]
图12-与非结合对照(2vhfc-va-eda-pnu)相比b1hfc-vc-pab-eda-pnu结合物在ror1敲除的pa-1细胞系中的效力。敲除ror1的pa-1细胞系中的b1hfc-vc-pab-eda-pnu结合物未显示出细胞杀伤。
[0146]
图13-tras(s442c)-vc-pab-eda-pnu结合物对her2阳性细胞系sk-br-3和her2阴性细胞系mda-mb-468的细胞杀伤数据。
[0147]
图14-tras(s442c)-va-eda-pnu结合物对her2阳性细胞系sk-br-3和her2阴性细胞系mda-mb-468的细胞杀伤数据。
[0148]
图15-p3a1hfc(s442c)-va-eda-pnu结合物对ror1阳性pa-1细胞系和ror1敲除的pa-1细胞系的杀伤效力。
[0149]
图16-多聚体vnar结合物与细胞表面ror1(a549癌细胞系)的结合。(a)ba11-b1-d3对ba11-b1-d3-pnu结合物;(b)p3a1-ba11-d3对p3a1-ba11-d3-pnu结合物;(c)p3a1-ba11-p3a1对p3a1-ba11-p3a1-pnu结合物。与vc-pab-eda-pnu或va-eda-pnu连接基团有效载荷结合后,维持与ror1的结合。
[0150]
图17-b1-hfc-vc-pab-eda-pnu和b1-hfc-va-eda-pnu在针对经载体处理小鼠的pdx胸膜间皮瘤模型中的体内疗效评估。绘制的绝对平均肿瘤体积 /-平均值标准误差(n=5)。在平均肿瘤体积为124mm3时开始治疗。在第1、4、7、10、18天通过静脉注射给予pdc分子治疗(用箭头表示);在第一次pdc给药前20小时,所有小鼠均预先由migg处理。载体数据绘制到最后一天,此时所有小鼠均存活并出现在该组中。
[0151]
图18-b1-hfc-vc-pab-eda-pnu和b1-hfc-va-eda-pnu在针对经载体处理小鼠的tnbc的pdx模型中的体内疗效评估。绘制的绝对平均肿瘤体积 /-平均值标准误差(n=5)。在平均肿瘤体积为180mm3时开始治疗。在第2、5、8、12、15天通过静脉注射给予pdc分子治疗
(用箭头表示);在第一次pdc给药前20小时,所有小鼠均预先由migg处理。载体数据绘制到最后一天,此时所有小鼠均存活并出现在该组中。
具体实施方式
[0152]
蒽环类药物是一类可用作药物结合物的有效载荷的引起高度关注的dna插入毒素,因为蒽环类药物已被临床验证是癌症治疗中的化疗药物。蒽环类衍生物pnu-159682被描述为奈莫柔比星的代谢物(quintieri等人(2005)clin.cancer res.11,1608-1617)且据报道对一个卵巢(a2780)细胞系和一个乳腺癌(mcf7)细胞系在皮摩尔至飞摩尔范围内的体外细胞杀伤表现出极高的效力(wo2012/073217a1)。
[0153]
化学结合蛋白-药物结合物的稳定性是一个重要的考虑因素,因为在靶向肿瘤细胞之前,患者循环中意外释放高效力蒽环类毒素(如pnu-159682)会导致靶外效应和不良副作用。图1给出了从pnu结合物释放的一些示例性分子,其显示了来自不同的含val-cit-pab的药物连接基团释放pnu159682和eda-pnu159682衍生物。
[0154]
因此,为了避免或至少减少不必要的副作用,需要能够与具有高稳定性的靶向蛋白连接的强效毒素。作为选择,连接基团有效载荷的设计使得细胞外裂解释放具有衰减效力的有效载荷衍生物。然而,需要保留足够的效力,以避免由于需要施用更高的剂量以达到疗效而导致的副作用的减少被抵消。
[0155]
易于结合是生产易于制造的产品的一个重要因素。本发明第一方面的有效载荷使用马来酰亚胺基团,其可使用直接和标准条件与结合伴侣上的任何可用硫醇基团反应。此外,使用马来酰亚胺/硫醇化学进行结合允许对引入的硫醇基团进行位点特异性结合,例如在蛋白质序列中的工程化半胱氨酸残基的侧链上。在本文描述的一些情况下,半胱氨酸可以通过在蛋白质的c-或n-末端引入含有工程化半胱氨酸(示例性序列包括但不限于qackahhhhhhgaefeqkliseedl(seq id no:52)或qacgahhhhhhgaefeqkliseedl(seq id no:53))的his myc标签来引入。
[0156]
使用非选择性标记方法(例如通过与蛋白质内的氨基官能团反应)生成的抗体/蛋白质-药物结合物提供含有多种不同物种的产品,具有不同的药物-抗体比率。这会影响结合物的性质,包括影响体内药效和毒性的效力和pk性质。因此,硫醇反应性有效载荷非常重要,因为它们可以在简单的过程中以高收率与蛋白质中天然存在的半胱氨酸残基反应,或与利用分子生物学/重组蛋白质表达或化学合成或通过对表达的、合成的或天然的蛋白质的化学修饰而加工到蛋白质的序列内任意点的特异性位点的半胱氨酸残基反应。
[0157]
本发明提供适用于药物结合物,包括但不限于蛋白质-药物结合物(pdcs)的蒽环类(pnu)衍生物。具体而言,提供了pnu159682的衍生物,其缺少c14碳和附着的羟基官能团,并且其中乙二胺(eda)基团形成pnu159682的c13羰基和马来酰亚胺基团之间的连接区域的一部分。马来酰亚胺基团存在于本发明第一方面的蒽环类(pnu)衍生物中,并且也可能存在于本发明第二方面的蒽环类(pnu)衍生物中。这种有效载荷能够与另一分子上的游离硫醇基团反应。当游离硫醇位于蛋白质上时,可形成蛋白质-药物结合物(pdc)。
[0158]
令人惊讶的是,与效力稍低的非eda有效载荷或释放的有效载荷衍生物相比,由乙二胺(eda)基团官能化并经由马来酰亚胺基团连接到硫醇基团的pnu159682衍生物显示出更高的稳定性。更稳定的有效载荷可能是有利的,因为减少了脱靶效应,这反过来可能会减
少副作用并提高患者的依从性。
[0159]
本发明进一步提供一种靶结合分子-药物结合物,其包括根据上述公开的蒽环类衍生物结合物和靶结合分子。
[0160]
根据靶结合分子-药物结合物的另一个实施方式,靶结合分子是蛋白质,并且蒽环类(pnu)衍生物选择性地通过一个或多个连接基团与引入蛋白质氨基酸序列中的硫醇基团结合。引入的硫醇可在蛋白质的氨基或羧基末端被引入,或引入其结构域或亚基的氨基或羧基末端。在另一个实施方式中,这种结合是与引入蛋白质的氨基或羧基末端的序列中的硫醇基团结合,或与其结构域或亚基的氨基或羧基末端结合。
[0161]
靶结合分子可以是蛋白质,例如特异性抗原结合蛋白可以是源自软骨鱼血清中发现的新颖的或新的抗原受体(ignar)的vnar结构域(greenberg a.s.,等人,nature,1995.374(6518):168-173页;dooley,h.,等人,mol.immunol,2003.40(1):25-33页;m
ü
ller,m.r.,等人,mabs,2012.4(6):673-685页)。
[0162]
因此,特异性抗原结合蛋白可包含由式(iii)表示的氨基酸序列:
[0163]
fw1-cdr1-fw2-hv2-fw3a-hv4-fw3b-cdr3-fw4
ꢀꢀꢀ
(iii)
[0164]
其中
[0165]
fw1是框架区
[0166]
cdr1是cdr序列
[0167]
fw2是框架区
[0168]
hv2是高变序列
[0169]
fw3a是框架区
[0170]
hv4是高变序列
[0171]
fw3b是框架区
[0172]
cdr3是cdr序列
[0173]
fw4是框架区。
[0174]
框架区fw1优选长度为20至28个氨基酸,更优选长度为22至26个氨基酸,进而更优选长度为23至25个氨基酸。在某些优选实施方式中,fw1的长度为26个氨基酸。在其他优选实施方式中,fw1的长度为25个氨基酸。在另外其他的优选实施方式中,fw1的长度是24个氨基酸。
[0175]
cdr区cdr1的长度优选为7至11个氨基酸,长度更优选为8至10个氨基酸。在某些优选实施方式中,cdr1的长度为9个氨基酸。在其他优选实施方式中,cdr1的长度为8个氨基酸。
[0176]
框架区fw2的长度优选为6至14个氨基酸,长度更优选为8至12个氨基酸。在某些优选实施方式中,fw2的长度为12个氨基酸。在其他优选实施方式中,fw2的长度为10个氨基酸。在其他优选实施方式中,fw2的长度为9个氨基酸。在其他优选实施方式中,fw2的长度为8个氨基酸。
[0177]
高变序列hv2的长度优选为4至11个氨基酸,长度更优选为5至10个氨基酸。在某些优选实施方式中,hv2的长度为10个氨基酸。在某些优选实施方式中,hv2的长度为9个氨基酸。在其他优选实施方式中,hv2的长度为6个氨基酸。
[0178]
框架区fw3a的长度优选为6至10个氨基酸,长度更优选为7至9个氨基酸。在某些优
选实施方式中,fw3a的长度为8个氨基酸。在某些优选实施方式中,fw3a的长度为7个氨基酸。
[0179]
高变序列hv4的长度优选为3至7个氨基酸,长度更优选为4至6个氨基酸。在某些优选实施方式中,hv4的长度为5个氨基酸。在其他优选实施方式中,hv4的长度为4个氨基酸。
[0180]
框架区fw3b的长度优选为17至24个氨基酸,长度更优选为18至23个氨基酸,长度进而更优选为19至22个氨基酸。在某些优选实施方式中,fw3b的长度为21个氨基酸。在其他优选实施方式中,fw3b的长度为20个氨基酸。
[0181]
cdr区cdr3的长度优选为8至21个氨基酸,长度更优选为9至20个氨基酸,长度进而更优选为10至19个氨基酸。在某些优选实施方式中,cdr3的长度为17个氨基酸。在其他优选实施方式中,cdr3的长度为14个氨基酸。在进而其他优选的实施方式中,cdr3的长度为12个氨基酸。在另外其他优选的实施方式中,cdr3的长度为10个氨基酸。
[0182]
框架区fw4的长度优选为7至14个氨基酸,长度更优选为8至13个氨基酸,长度进而更优选为9至12个氨基酸。在某些优选实施方式中,fw4的长度为12个氨基酸。在其他优选实施方式中,fw4的长度为11个氨基酸。在进而其他优选的实施方式中,fw4的长度为10个氨基酸。在另外其他优选的实施方式中,fw4的长度为9个氨基酸。
[0183]
本文明确考虑上述框架区、互补决定区和高变区的所有可能组合和排列。
[0184]
用于本发明的优选vnar结构域包括b1、p3a1、d3、ba11和e9,其序列如下所示。b1、p3a1、d3和e9结合至ror1(数据显示在待审国际专利申请号pct/ep2018/086823中,作为wo 2019/122447公开,其内容以引用的方式并入本文)pct。ba11是以高亲和力结合人血清白蛋白的人源化vnar(kovalenko等人,j.biol.chem.,2013jbc)。另外,下面还描述了非结合vnar结构域2v。
[0185]
b1是asvnqtprtatketgesltincvvtganyglaatywyrknpgssnqerisisgryvesvnkrtmsfslrikdltvadsatyyckaypwgagapwlvqwydgagtvltvn(seq id no:44)
[0186]
2v是trvdqtprtatketgesltincvltdtsyglystswfrknpgttdwermsiggryvesvnkgaksfslrikdltvadsatyyckaqslaistrsywydgagtvltvn(seq id no:54)
[0187]
p3a1是trvdqtprtatketgesltincvltdtsyglystswfrknpgttdwermsiggryvesvnkgaksfslrikdltvadsatyyckarearhpwlrqwydgagtvltvn(seq id no:43)
[0188]
d3是asvnqtprtatketgesltincvltdtsyglystswfrknpgttdwermsiggryvesvnkraksfslrikdltvadsatyyckaqsgmaistgsghgynwydgagtvltvn(seq id no:39)
[0189]
ba11是trvdqspsslsasvgdrvtitcvltdtsyplystywyrknpgssnkeqisisgrysesvnkgtksftltisslqpedsatyycramstniwtgdgagtkveik(seq id no:55)
[0190]
e9是akvdqtprtatketgesltincvltdtsyglystswfrknpgttdwermsiggryvesvnkraksfslrikdltvadsatyyckaqsgmaidigsghgynwydgagtvltvn(seq id no:40)
[0191]
用于本发明的vnar结构域包括seq id nos:40-49和59中标识的vnar。其它优选的vnars包括seq id nos:45-49和59中标识的vnars。特别优选的人源化vnar是b1v15,具有氨基酸序列asvtqsprsasketgesltitcrvtganyglaatywyrknpgssnqerisisgrysesvnkrtmsfslrissltvedsatyyckaypwgagapwlvqwydgqgtklevk(seq id no:59)。
[0192]
与本发明的分子相关的序列同一性可在单个cdr、hv或fw的水平上判断,或可在整个分子的长度上进行判断。所述的cdr、hv和fw序列也可以更长或更短,无论是通过在序列
的n末端或c末端添加或删除氨基酸,还是通过插入或删除带有序列的氨基酸。
[0193]
特异性结合蛋白的任何部分可被设计以实现本发明pdc中的结合。在优选的实例中,在使用免疫球蛋白fc区的情况下,可将其设计为包含半胱氨酸残基作为结合位点。优选的引入的半胱氨酸包括但不限于s252c和s473c(kabat编号),分别对应于eu编号中的s239c和s442c。
[0194]
靶结合分子-药物结合物可以是本文公开的任何靶结合分子-药物结合物。例如,靶结合分子-药物结合物可选自由由以下组成的组:
[0195]
b1-hfc-vc-pab-eda-pnu
[0196]
b1-hfc-va-eda-pnu
[0197]
b1-hfc-na-eda-pnu
[0198]
b1-hfc-vc-pab-eda-pnu和b1-hfc-va-eda-pnu在本文中显示出在治疗间皮瘤和tnbc中具有特殊功效。
[0199]
优选地,靶结合分子-药物结合物包含peg4间隔基。例如,靶结合分子-药物结合物可选自由以下组成的组:
[0200]
b1-hfc-peg4-vc-pab-eda-pnu
[0201]
b1-hfc-peg4-va-eda-pnu
[0202]
b1-hfc-peg4-na-eda-pnu
[0203]
优选地,hfc包含s239c(eu编号)处引入的半胱氨酸残基。因此,例如,靶结合分子-药物结合物可选自由以下组成的组:
[0204]
b1-hfc(s239c)-vc-pab-eda-pnu
[0205]
b1-hfc(s239c)-va-eda-pnu
[0206]
b1-hfc(s239c)-na-eda-pnu
[0207]
靶结合分子-药物结合物可选自由以下组成的组:
[0208]
b1-hfc(s239c)-peg4-vc-pab-eda-pnu
[0209]
b1-hfc(s239c)-peg4-va-eda-pnu
[0210]
b1-hfc(s239c)-peg4-na-eda-pnu
[0211]
在靶结合分子-药物结合物的一个实施方式中,靶结合分子是抗体。在靶结合分子-药物结合物的另一个实施方式中,靶结合分子结合her-2。优选地,靶结合分子是对her-2具有特异性的抗体。
[0212]
优选地,靶结合分子-药物结合物包含peg4间隔基。例如,靶结合分子-药物结合物可选自由以下组成的组:
[0213]
tras-peg4-vc-pab-eda-pnu
[0214]
tras-peg4-va-eda-pnu
[0215]
靶结合分子-药物结合物可选自由以下组成的组:
[0216]
tras(s442c)-peg4-vc-pab-eda-pnu
[0217]
tras(s442c)-peg4-va-eda-pnu
[0218]
关于本发明的任何上述方面所描述的任何特征可在必要时与本发明的其他方面相结合。
[0219]
定义
[0220]
如本文所用,烷基基团是含有1至40个碳原子的直链或支链、被取代或未被取代的基团(优选未被取代)。烷基基团可任选地在任何位置被取代。如本文所使用的术语“烯基”表示从具有至少一个碳-碳双键的直链或支链脂肪族部分去除单个氢原子而衍生的基团。如本文所使用的术语“炔基”是指从具有至少一个碳-碳三键的直链或支链脂肪族部分去除单个氢原子而衍生的基团。
[0221]
术语
‘
烷基’、
‘
芳基’、
‘
杂芳基’等还包括多价物种,例如亚烷基、亚芳基、
‘
杂亚芳基’等。亚烷基基团的实例包括乙烯基(-ch
2-ch
2-)和丙烯基(-ch
2-ch
2-ch
2-)。示例性亚芳基基团是亚苯基(-c6h
4-),示例性杂亚芳基基团是吡啶亚基(-c5h3n-)。
[0222]
芳环是可具有0、1、2或更多、优选0、1或2个环状杂原子的环状芳基。芳环可任选地被取代和/或可与一个或多个芳环或非芳环(优选芳环)稠合,其可包含0、1、2或多个环状杂原子,以形成多环系统。
[0223]
芳环包括芳基和杂芳基基团二者。芳基和杂芳基基团可以是单核的,即只有一个芳环(例如像苯基或亚苯基),或多核的,即有两个或更多个可稠合的芳环(例如像萘基或亚萘基),单独共价连接(例如像联苯)和/或稠合和单独连接的芳环二者的组合。优选地,芳基或杂芳基基团是在基本上整个基团上基本上共轭的芳族基团。芳基基团可包含5至40个环碳原子、5至25个环碳原子、5至20个环碳原子或5至12个环碳原子。杂芳基基团可为5至40元、5至25元、5至20元或5至12元环,包含1个或多个选自n、o、s和p的环状杂原子。芳基或杂芳基可稠合至一个或多个芳环或非芳环(优选芳环)以形成多环系统。
[0224]
芳基和杂芳基优选地表示具有最多25个环原子的单环、双环或三环芳族基团或杂芳族基团,其也可包含稠环且任选地被取代。
[0225]
优选的芳基基团包括但不限于苯、联苯、苯并菲、[1,1':3',1”]三联苯-2'-亚基、萘、蒽、联亚萘基、菲、二氢芘、二萘嵌苯、并四苯、并五苯、苯并芘、芴、茚、茚并芴、螺双芴等。
[0226]
优选的杂芳基基团包括但不限于5元环,如吡咯、吡唑、噻咯、咪唑、1,2,3-三唑、1,2,4-三唑、四唑、呋喃、噻吩、硒酚、噁唑、异噁唑、1,2-噻唑、1,3-噻唑、1,2,3-噁二唑、1,2,4-噁二唑、1,2,5-噁二唑、1,3,4-噁二唑、1,2,3-噻二唑、1,2,4-噻二唑、1,2,5-噻二唑、1,3,4-噻二唑;6元环,如吡啶、哒嗪、嘧啶、吡嗪、1,3,5-三嗪、1,2,4-三嗪、1,2,3-三嗪、1,2,4,5-四嗪、1,2,3,4-四嗪、1,2,3,5-四嗪;以及稠合系统,如咔唑、吲哚、异吲哚、吲嗪、吲唑、苯并咪唑、苯并三唑、嘌呤、萘并咪唑、菲并咪唑、吡啶并咪唑、吡嗪并咪唑、喹喔啉并咪唑、苯并噁唑、萘并噁唑、蒽并噁唑、菲并噁唑、异噁唑、苯并噻唑、苯并呋喃、异苯并呋喃、二苯并呋喃、喹啉、异喹啉、蝶啶、苯并-5,6-喹啉、苯并-6,7-喹啉、苯并-7,8-喹啉、苯并异喹啉、吖啶、吩噻嗪、吩噁嗪、苯并哒嗪、苯并嘧啶、喹喔啉、二氮蒽、二氮杂萘、氮咔唑、苯并咔啉、菲啶、菲咯啉、噻吩并[2,3b]噻吩、噻吩并[3,2b]噻吩、二噻吩并噻吩、二噻吩并吡啶、异苯并噻吩、二苯并噻吩、苯并噻二唑并噻吩、2,5-二氢吡咯[3,4-c]吡咯-1,4-二酮(吡咯并吡咯二酮,dpp)、2-氧代-1h-吲哚-3-亚基、[3,3'-双吡咯[2,3-b]亚吡啶]-2,2'(1h,1'h)-二酮(吡啶异靛蓝)和(3e)-3-(2-氧代-1h-吲哚-3-亚基)-1h-吲哚-2-酮(异靛蓝)或其组合。杂芳基基团可被烷基、烷氧基、硫烷基、氟、氟烷基或其它芳基或杂芳基取代基取代。优选地,杂芳基基团为噻吩。
[0227]
特别优选的杂原子选自o、s、n、p和si。通常,氢将完成本发明分子中包含的杂原子
的价态,例如,对于n,可能存在-nh-或-nh2,其中涉及一个或两个其他基团。
[0228]
如本文所用,术语“任选取代”意指任选取代部分中的一个或多个氢原子被合适的取代基取代。除非另有说明,“任选取代”基团可在该基团的每个可取代位置处具有合适的取代基,并且当任何给定结构中的多于一个位置可被选自指定基团中的多于一个取代基取代时,取代基在每个位置处可为相同或不同。本发明设想的取代基的组合优选是导致形成稳定化合物的那些。本文使用的术语“稳定”是指化学上可行且在室温(即16℃至25℃)下能够存在足够长时间以允许其检测、分离和/或用于化学合成的化合物。
[0229]
上述任何基团(例如,本文中称为“任选取代”的基团,包括烷基、芳基和杂芳基)可任选地包括一个或多个取代基,优选选自甲硅烷基、磺基、磺酰基、甲酸基、氨基、亚氨基、次氨基、巯基、氰基、硝基、卤素、-nco、-ncs、-ocn、-scn、-c(=o)nr0r
00
、-c(=o)x0、-c(=o)r0、-nr0r
00
、c
1-12
烷基、c
1-12
烯基、c
1-12
炔基、c
6-12
芳基、c
3-12
环烷基、具有4至12个环原子的杂环烷基、具有5至12个环原子的杂芳基、c
1-12
烷氧基、羟基、c
1-12
烷基羰基、c
1-12
烷氧基-羰基、c
1-12
烷基羰氧基或c
1-12
烷氧基羰氧基,其中一个或多个h原子任选地被f或cl和/或其组合取代;其中x0是卤素,且r0和r
00
独立地为h或任选取代的c
1-12
烷基。任选取代基可包括同一基团和/或多个前述基团所有化学上可能的组合(例如,如果直接彼此连接氨基和磺酰基表示氨磺酰自由基)。在一个实施方式中,取代基不是酰基。如本文所用,酰基是指酰基基团,其是通过从含氧酸(例如羧酸)去除一个或多个羟基基团而衍生的部分。它包含双键氧原子和烷基基团。
[0230]
在一些实施方式中,基团可以未被取代。例如,关于本发明的第一方面,蒽环类(pnu)衍生物可为式(i):
[0231][0232]
其中[x]是选自包括未被取代的烷基基团、未被取代的杂烷基基团、未被取代的芳基基团、未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基;
[0233]
[l1]和[l2]是选自由缬氨酸(val)、瓜氨酸(cit)、丙氨酸(ala)、天冬酰胺(asn)、肽、-(ch2)n-、-(ch2ch2o)
n-、对氨基苄氧基羰基(pab)、val-cit-pab、val-ala-pab、ala-ala-asn-pab、除甘氨酸外的任何氨基酸及其组合组成的组的任选连接基团。
[0234]
在基团未被取代的实施方式中,[x]优选选自包括聚乙二醇和的
组,其中表示与分子的其余部分连接的点并且其中[r]是选自包括未被取代的烷基基团、未被取代的杂烷基基团、未被取代的芳基基团、未被取代的杂芳基基团、一种或多种杂原子、聚乙二醇或其组合的组的任选间隔基。
[0235]
一般而言,术语pab意指对氨基苄氧基羰基。偶尔在文献中,术语pab可用于表示对氨基苄基。在本说明书中,pab旨在表示对氨基苄氧基羰基。
[0236]
术语“蛋白质”通常指通过肽键连接在一起的多个氨基酸残基。它与肽、寡肽、寡聚物或多肽的含义相同并且可以互换使用,并包括糖蛋白及其衍生物。术语“蛋白质”还旨在包括蛋白质的片段、类似物、变体和衍生物,其中片段、类似物、变体或衍生物保留与参考蛋白基本相同的生物活性或功能。蛋白质类似物和衍生物的实例包括肽核酸和darpins(设计的锚蛋白重复蛋白)。
[0237]
术语“靶结合分子”是指与给定靶结合的任何分子。在这种情况下,“靶”和“抗原”可互换使用。靶结合分子的实例包括天然或重组蛋白质,包括免疫球蛋白或抗体、免疫球蛋白fc区、免疫球蛋白fab区、fab’、fv、fv-fc、单链fv(scfv)、scfv-fc、(scfv)2、双特异抗体、三特异抗体、四特异抗体、双特异性t-细胞结合器(bite)、内含肽、内含肽融合、vnar结构域、单结构域抗体(sdab)、vh结构域、支架蛋白质(亲和体、centyrin、darpin等),以及核酸,该核酸包括已开发用于结合靶或自然结合靶的适体或小分子或天然产物。
[0238]
抗原特异性结合蛋白可包括与给定抗原结合的任何蛋白。优选实例包括免疫球蛋白或抗体、免疫球蛋白fc区、免疫球蛋白fab区、fab’、fv、fv-fc、单链fv(scfv)、scfv-fc、(scfv)2、双特异抗体、三特异抗体、四特异抗体、双特异性t-细胞结合器(bite)、内含肽、内含肽融合、vnar结构域、单结构域抗体(sdab)、vh结构域或支架蛋白质(亲和体、centyrin、darpin等)。特别优选的实例包括vnar结构域,其包含源自vnar分子合成文库或源自软骨鱼免疫的文库的氨基酸序列。术语vnar、ignar和nar也可互换使用。
[0239]
氨基酸在本文中表示为单字母代码或三字母代码或两者。
[0240]
蛋白质和生物分子经化学修饰引入硫醇已被广泛证实。方法包括胺基与2-亚氨基硫烷(traut试剂)反应,用含有nhs-酯的异双功能试剂(如n-琥珀酰亚胺s-乙酰硫代酯(sata)或n-琥珀酰亚胺-4-(2-吡啶基二硫代)丁酸酯(spdb))修饰胺基,然后分别用羟胺和还原剂处理,并用半胱胺切割工程化的内含肽-融合蛋白以生成c末端巯基蛋白质和肽。
[0241]
术语“亲和纯化”是指基于分子对化学或结合伴侣的特定吸引或结合来纯化分子以形成组合或络合物,这允许分子与杂质分离,同时保持与伴侣部分的结合或吸引。
[0242]
术语“互补决定区”或cdr(即cdr1和cdr3)是指vnar结构域的氨基酸残基,其存在通常涉及抗原结合。每个vnar通常有两个cdr区,分别标识为cdr1和cdr3。另外,每个vnar结构域包含来自“高变环”(hv)的氨基酸,其也可能涉及抗原结合。在一些情况下,互补决定区可包含来自cdr区和高变环二者的氨基酸。在其他情况下,抗原结合可能仅涉及来自单个cdr或hv的残基。根据公认的vnar分子命名法,cdr2区不存在。
[0243]“框架区”(fw)是指除cdr残基以外的vnar残基。每个vnar通常有五个框架区,标识为fw1、fw2、fw3a、fw3b和fw4。
[0244]
vnar中fw、cdr和hv区域之间的边界不是固定的,因此,预计这些区域的长度和组成会发生一些变化。本领域技术人员将理解这一点,特别是关于在分析这些区域中已经执行的工作。(anderson等人,plos one(2016)11(8);lui等人,mol immun(2014)59,194-199;
zielonka等人,mar biotechnol(2015),17,(4)386-392;fennell等人,j mol biol(2010)400,155-170;kovalenko等人,j biol chem(2013)288,17408-17419;dooley等人,(2006)pnas 103(6),1846-1851)。尽管本发明的分子在本文中通过参考fw、cdr和hv区域来定义,但不限于这些严格的定义。因此,本文明确考虑与本领域中vnar结构域结构的理解一致的变化。
[0245]“密码子集”是指一组不同的核苷酸三联体序列,用于编码所需的变异氨基酸。一组寡核苷酸,例如,可通过固相合成来合成,包括表示由密码子集提供的核苷酸三联体的所有可能组合并将编码所需氨基酸组的序列。密码子指定的标准形式是iub代码的形式,这在本领域中是已知的,并在本文中描述。
[0246]
密码子集通常由3个斜体大写字母表示,例如nnk、nns、xyz、dvk等。因此,“非随机密码子集”是指编码部分、优选完全满足本文所述氨基酸选择标准的精选氨基酸的密码子集。在该领域,例如trim方法中,在特定位置具有选定核苷酸“简并度(degeneracy)”的寡核苷酸的合成是众所周知的(knappek等人;j.mol.biol.(1999),296,57-86);garrard&henner,gene(1993),128,103)。此类具有特定密码子集的寡核苷酸组可使用商业核酸合成器(例如可从加利福尼亚州福斯特市应用生物系统公司获得)合成,或可在商业上获得(例如,从马里兰州罗克维尔的生命技术公司获得)。合成的具有特定密码子集的一组寡核苷酸通常包括具有不同序列的多个寡核苷酸,其差异由整个序列内的密码子集确定。根据本发明使用的寡核苷酸具有允许与vnar核酸模板杂交的序列,并且在方便的情况下还可以包含限制性内切酶位点。
[0247]“细胞”、“细胞系”和“细胞培养物”可互换使用(除非上下文另有说明),此类名称包括细胞或细胞系的所有后代。因此,例如,像“转化体”和“转化细胞”的术语包括原代受试细胞和由此衍生的培养物,而不考虑转移的数量。还被理解的是,由于有意或无意的突变,所有后代的dna含量可能并不完全相同。包括与在最初转化细胞中筛选的具有相同功能或生物活性的突变后代。
[0248]
特定分析中化学实体的“检测限”是该实体在该分析背景水平以上可检测到的最低浓度。例如,在噬菌体elisa中,显示特定抗原结合片段的特定噬菌体的“检测限”是特定噬菌体产生高于由未显示抗原结合片段的对照噬菌体产生的elisa信号的噬菌体浓度。
[0249]“融合蛋白”和“融合多肽”是指具有两个共价连接在一起的部分的多肽,其中每一部分是具有不同性质的多肽。该性质可以是生物性质,例如体外或体内活性。该性质也可以是简单的化学或物理性质,例如与目标抗原的结合、反应的催化等。两部分可通过单个肽键直接连接,或通过含有一个或多个氨基酸残基的肽连接基团连接。通常,这两个部分和连接基团彼此将处于阅读框架中。优选地,多肽的两部分获自异源或不同的多肽。
[0250]
本文中的术语“融合蛋白”通常指通过化学手段(包括氢键或盐桥),或通过蛋白质合成的肽键,或二者而结合在一起的一种或多种蛋白质。通常,融合蛋白将通过dna重组技术制备,并可在本文中称为重组融合蛋白。
[0251]“同一性”描述两个或更多个多肽序列或两个或更多个多核苷酸序列之间的关系,通过比较序列来确定。同一性还指多肽或多核苷酸序列(视情况而定)之间的序列相关度(同源性),由此类序列的字符串之间的匹配确定。尽管存在许多方法来测量两个多肽或两个多核苷酸序列之间的同一性,但通常用于确定同一性的方法被编入计算机程序中。确定
两个序列之间的同一性的优选计算机程序包括但不限于gcg程序包(devereux等人,nucleic acids research,12,387(1984),blastp,blastn和fasta(atschul等人,j.molec.biol.(1990)215,403)。
[0252]
优选地,使用hgmp(人类基因组绘图项目)提供的blast计算机程序(atschul等人,j.mol.biol.(1990)215,403-410)的默认参数,在氨基酸水平上,蛋白质的氨基酸序列与本文公开的氨基酸序列具有至少45%的同一性。
[0253]
更优选地,在氨基酸水平上,蛋白质序列可具有与本文所示的氨基酸序列为至少45%、46%、47%、48%、49%、50%、55%、60%、65%、66%、67%、68%、69%、70%、75%、80%、85%、90%且进而更优选95%(进而更优选至少96%、97%、98%或99%)的同一性。
[0254]
使用hgmp提供的blast计算机程序的默认参数,蛋白质还可包含具有与本文公开的序列为至少45%、46%、47%、48%、49%、50%、50%、55%、60%、65%、66%、67%、68%、69%、70%、75%、80%、85%、90%、95%、96%、97%、98%或99%同一性的序列。
[0255]“突变”是相对于参考核苷酸序列(如野生型序列)的一个或多个核苷酸的缺失、插入或替换。
[0256]“天然”或“自然产生”的vnar是指从非合成来源,例如从体外获得的组织来源,或从弹性鳃亚纲动物的血清中识别的vnar。这些vnar可包含在任何类型的免疫反应(无论是自然的还是诱导的)中产生的vnar。天然vnar包含氨基酸序列和构成或编码这些抗体的核苷酸序列。如本文所使用的,天然vnar不同于“合成vnar”,合成vnar指已从源或模板序列改变的vnar序列,例如,通过在特定位置用不同氨基酸替换、删除或添加一个或多于一个的氨基酸,该不同氨基酸提供不同于源抗体序列的抗体序列。
[0257]
蛋白质的片段、类似物、变体或衍生物的长度可至少为25个,优选为30或40个,或最多为50或100个,或60至120个氨基酸,取决于衍生其的原始蛋白质序列的长度。在某些情况下,90至120、100至110个氨基酸的长度可能是方便的。
[0258]
蛋白质的片段、衍生物、变体或类似物可以是(i)其中一个或多个氨基酸残基被保守或非保守氨基酸残基(优选地,保守氨基酸残基)取代的片段、衍生物、变体或类似物,并且这种被取代氨基酸残基可以是也可以不是由遗传密码编码的片段,或(ii)其中氨基酸残基中的一个或多个包含取代基的片段、衍生物、变体或类似物,或(iii)其中附加氨基酸与成熟多肽融合的片段、衍生物、变体或类似物,例如用于纯化多肽的先导序列或辅助序列。此类片段、衍生物、变体和类似物被视为在本领域技术人员根据本文教导的范围内。
[0259]“寡核苷酸”是短长度、单链或双链多脱氧核苷酸,通过已知方法(如磷酸三酯、亚磷酸酯或亚磷酰胺化学合成,使用固相技术)化学合成。如果基因的整个核酸序列已知,或与编码链互补的核酸序列可用,则进一步的方法包括使用聚合酶链反应(pcr)。作为选择,如果目标氨基酸序列已知,则可以使用每个氨基酸残基的已知和优选编码残基来推断潜在的核酸序列。寡核苷酸可在聚丙烯酰胺凝胶或分子筛柱上纯化或通过沉淀纯化。当dna与非核酸杂质(可能是极性、非极性、离子等)分离时,dna被“纯化”。
[0260]
起始或参考多肽(例如,源vnar或其cdr)的“变体”或“突变体”,例如,融合蛋白(多肽)或异源多肽(噬菌体异源)是多肽,其(1)具有不同于起始或参考多肽的氨基酸序列且(2)通过自然或人工诱变从起始或参考多肽衍生。此类变体包括,例如,关注的多肽氨基酸序列内残基的缺失和/或插入和/或替换。例如,使用包含非随机密码子集的寡核苷酸生成
的本发明的融合多肽将是相对于源vnar或抗原结合片段的变异多肽,该密码子集编码具有变异氨基酸的序列(相对于在源vnar或抗原结合片段的相应位置发现的氨基酸)。因此,变体cdr是指包含相对于起始或参考多肽序列(例如源vnar或抗原结合片段的序列)的变体序列的cdr。在该上下文中,变异氨基酸是指与起始或参考多肽序列(例如源vnar或抗原结合片段的序列)中对应位置处的氨基酸不同的氨基酸。只要最终结构具有所需的功能特性,则可进行任何删除、插入和替换的组合以获得最终变体或突变体结构。氨基酸的变化也可能改变多肽的翻译后过程,例如改变糖基化位点的数量或位置。
[0261]“野生型”或“参考”序列或“野生型”或“参考”蛋白质/多肽的序列,例如外壳蛋白或源vnar的cdr,可以是通过引入突变从中衍生出变异多肽的参考序列。一般来说,给定蛋白质的“野生型”序列是自然界中最常见的序列。类似地,“野生型”基因序列是自然界中最常见的基因序列。突变可通过自然过程或人类诱导的方式引入“野生型”基因(以及它编码的蛋白质)。这些过程的产物是原始“野生型”蛋白质或基因的“变体”或“突变体”形式。
[0262]
本文使用的术语“结合”可指化学连接两个或更多个化学部分的任何方法。通常,结合将通过共价键进行。在本发明的上下文中,至少一个化学部分将是靶结合分子,另一个或多个分子将是本发明的pnu衍生物。在一些情况下,除了本发明的pnu衍生物外,结合将涉及两个或更多个靶结合分子,在这种情况下,结合可直接在靶结合分子之间进行,其中pnu衍生物结合至一个靶结合分子。
[0263]
短语“选自包括
……
的组”可替换为短语“选自由
……
组成的组”,反之亦然,无论它们在本文中出现在何处。
[0264]
将通过参考以下实施例来进一步理解本发明。
[0265]
实施例
[0266]
实施例1:pnu衍生物的表征
[0267]
根据标准合成方法制备本发明的pnu衍生物。使用质谱法来验证是否产生了正确的分子(表1)。
[0268]
表1:pnu衍生物的质谱表征
[0269]
[0270][0271]
在多种细胞系中测试了eda-pnu159682衍生物的效力(图1)。结果汇总在表2中。
[0272]
表2:eda pnu159682衍生物游离有效载荷96小时的ic50
[0273]
[0274][0275]
实施例2:vnar-hfc pnu结合物
[0276]
为了研究本发明的pnu衍生物,制备了许多vnar-hfc-pnu结合物。两个vnar对ror1(b1和p3a1)具有特异性。另外,非结合的vnar(2v)用作对照分子。
[0277]
vnar通过标准[g4s]3基因融合到工程化的higg1 fc结构域,该higg1 fc结构域在higg1 fc序列s239c(eu编号)中含有半胱氨酸替代物。vnar fc融合蛋白在cho k1细胞中以分泌蛋白的形式表达,并使用mabselect
tm sure
tm
(evitria,瑞士)从培养基纯化。纯化蛋白通过sec(advancebio,agilent)、sds-page和质谱分析以确认序列和蛋白质完整性。使用pioneer surface plasmon resonance(spr)设备(sensiq/pall fortebio)或biolayer interferometry(bli)octet k2系统(fortebio)测定结合动力学。ror1-hfc或ror2-hfc融合蛋白(胞外域)在乙酸钠ph 5缓冲液中使用胺偶联固定至cooh2芯片或ar2g传感器。在不同浓度下测试vnar和vnar fc分子,并使用qdat软件(sensiq/pall fortebio)或用于生物层干涉测量的八位组数据分析高通量软件(fortebio)测定ka(m-1
s-1
)、kd(s-1
)和kd(nm)值。包括ror1 2a2 mab(biolegend)和ror2 mab(r&d systems)作为与ror1和ror2的阳性/阴性结合的对照。2v是对照vnar序列,来源于原始的vnar文库,因此是该蛋白质类的代表,但没有已知的靶点。表3和3b总结了这些分子对人ror1和人ror2亲和力的表面等离子体共振数据。
[0278]
表3-vnar fc融合与人ror1和人ror2结合的spr数据
[0279][0280]
对p3a1 hfc(442)重复相同的过程,得到可比较的结合数据。对于该衍生物,vnar基因融合至工程化higg1 fc结构域,该结构域在higg1 fc序列s442c(eu编号)中含有半胱氨酸替代物。该分子对人ror1和人ror2亲和力的表面等离子体共振数据如表3b所示。
[0281]
表3b-vnar fc融合与人ror1和人ror2结合的spr数据
[0282]
[0283][0284]
采用改编自文献[junutula等人,2008nat biotech;jeffrey等人,2013bioconj chem]的部分还原、重折叠和标记方法,这些蛋白质用马来酰亚胺pnu衍生物进行了位点特异性标记(图1和2)。简而言之,在pbs 100mm l-精氨酸ph 7.4与1mm edta中制备1mg/ml vnar hfc溶液。添加20摩尔当量tcep,并在4℃孵育至少48小时。添加30摩尔当量dhaa,ph调整至6.5,并在室温下孵育1小时。在室温下与4或5摩尔当量的马来酰亚胺pnu溶液反应过夜之前,将重新折叠的vnar fc s239c广泛透析或缓冲液交换成pbs 50mm l-精氨酸,并通过uv进行定量。结合物通过sec纯化,并通过分析hic、分析sec和lc-ms进行分析。表4总结了制备的结合物。
[0285]
表4:vnar-pnu结合物的特征概述
[0286]
[0287][0288]
对p3a1 hfc(442)-va-eda-pnu结合物重复相同的过程。表4b总结了制备的结合物。
[0289][0290]
抗ror1-vnar药物结合物治疗癌细胞的体外细胞活性测定
[0291]
将细胞接种到白色透明底部的96孔板(costar)中,并在37℃、5%co2下孵育24小时。第二天,以x10工作原液为每种试验剂建立稀释系列。x10原液的剂量反应为:10000、5000、1000、500、100、50、10、5、1、0.5nm等。使用多通道移液管将10μl x10原液添加至细胞板(每孔90μl)。这导致在孔中1:10的稀释,对于最敏感的细胞系,剂量反应范围从1000nm(第1列)到0.05nm(第10列)或持续到0.5fm(如果需要)。将10μl载体对照(pbs)添加到对照孔(第11列和第12列)中。板在37℃、5%co2下孵育72至96小时。按照制造商的说明使用promega cell titre glo试剂评估细胞活性。简而言之,将分析板从培养箱中取出并使其平衡至室温,然后向每个100μl分析孔中添加100μl室温cell titre glo试剂。将板放置在板式振动筛上,以600rpm振动2分钟。允许平板在室温下再放置10分钟,之后使用clariostar平板读取器(bmg)测量发光读数。通过计算未处理(仅载体)对照孔的平均值并确定每个处理孔的对照百分比来分析数据。然后使对照数据的百分比相对于log[处理]浓度绘图,使用graphpad prism软件中的非线性回归拟合得出ic50值。
[0292]
使用以下细胞系:
[0293]
kasumi-2-人b细胞白血病前体;
[0294]
pa-1-人卵巢癌细胞系;
[0295]
pa-1ror1 ko-敲除ror1的人卵巢癌细胞系;
[0296]
697-人b细胞白血病前体,和
[0297]
mhh-es1-人尤文肉瘤细胞系。
[0298][0299]
表6:ror1特异性结合物与非结合2v hfc结合物的比较
[0300][0301]
表5和6以及图3至图12和图15显示,使用本发明有效载荷的ror1靶蛋白质药物结合物以ror1依赖的方式高度有效地杀死ror1表达癌细胞,与相应的非结合蛋白质药物结合物(2v hfc)相比具有较大的窗口。
[0302]
表7:非结合vnar(2v)-hfc-pnu159682结合物的效力
[0303]
2v是对照vnar序列,来源于原始的vnar文库,因此不具有已知的靶点,也不会通过流式细胞术与癌细胞系结合。如前所述,使用一组癌细胞系生成2v hfc-pnu结合物并评估其非选择性细胞杀伤。
[0304][0305][0306]
表7中的数据表明,本发明的pnu结合物始终比现有技术的peg4-vc-pab-dmae-pnu159682具有更高的ic50值。因此,本发明的连接基团-有效载荷产生更稳定的结合物和/或更低效的副产物,并且这些蛋白质-药物结合物对正常组织的毒性应更小。
[0307]
实施例3:曲妥珠单抗结合物
[0308]
产生曲妥珠单抗突变体,其中fc部分(位置442)中的单个丝氨酸残基变为半胱氨酸残基。这产生曲妥珠单抗分子,其在fc部分内重链的442位置具有独特的硫醇用于结合。此类突变体可称为曲妥珠单抗s442c或tras(s442c)。
[0309]
新型pnu有效载荷中的两个使用上述方法通过工程化半胱氨酸与该分子结合,以良好的总收率提供相应的结合物。下表8概述了这些结合物的性质。
[0310]
表8:tras(s442c)-pnu结合物的分析
[0311][0312]
此外,研究了tras(s442c)-pnu结合物对her2阳性细胞系的杀伤效力。图13和14显示两种tras(s442c)-pnu结合物均选择性杀死her2阳性细胞系sk-br-3,对her2阴性细胞系mda-mb-468几乎没有影响。
[0313]
实施例4:vnar-pnu结合物
[0314]
多聚体和双特异性vnar结构由用于位点特异性标记的含有工程化半胱氨酸的c末端his-myc标签生成。蛋白质用2mm tcep处理,并用imac纯化。
[0315]
如前所述,使用pioneer表面等离子体共振(spr)仪器(sensiq/pall fortebio)或生物层干涉仪(bli)octet k2系统(fortebio)测定多聚体vnar蛋白与ror1hfc或ror2 hfc融合蛋白(胞外域)结合的结合动力学。
[0316]
表9:多聚体vnars与人ror1和人ror2结合的spr数据
[0317][0318]
使用4当量pnu在室温下进行结合1小时,并通过sec纯化。通过分析hic、分析sec和lc-ms分析最终结合物。
[0319]
表10:多聚的和双特异性结合物的表征
[0320][0321]
vnar结构域之间的连接基团是(g4s)5[用([g4s]5]表示;pgvqpspggggs[用(wbg4s)表示](seq id no:50);pgvqpapggggs[用(wbg4sgm)表示](seq id no:51)。
[0322]
有效载荷通过引入含有工程化半胱氨酸(序列为qackahhhhhhgaefeqkliseedl(seq id no:52)或qacgahhhhhhgaefeqkliseedl(seq id no:53))的c-末端his-myc标签与蛋白质c-末端区引入的独特游离硫醇结合。
[0323]
流式细胞术检测多聚体结合物与ror1
hi a549肺腺癌细胞表面的结合。通过在37℃下用0.1%edta/pbs溶液孵育约10分钟从组织培养瓶中分离出粘附的人类癌细胞或直到细胞容易分离。细胞重新悬浮在15ml试管中5ml冰冷pbs/2%fcs中,并在4℃以1500rpm离心5分钟。去除上清液,细胞颗粒重新悬浮在1ml至2ml的pbs/2%fcs中。使用z1 coulter粒子计数器(beckman coulter)进行细胞计数,并将每个试样的5x 10^5个细胞等分到96孔板中。将细胞与100μl指定浓度的vnar(his6myc标记)或相应的vnar结合物以及对照在冰上孵育1小时。样品板以2000rpm下离心5分钟。去除上清液,并通过使用多通道移液管将细胞颗粒重新悬浮在0.25ml冰冷pbs/2%fcs中进行洗涤。样品再次在4℃以2000rpm离心5分钟。去除上清液,并按照所述进行两次进一步洗涤。在最后的洗涤和离心步骤后,通过在薄纸上吸干板去除多余的液体。每个细胞颗粒添加100μl抗x6his tag ab(abcam),以适当地结合vnar(his6myc标记)或相应的vnar结合物,并在冰上孵育30分钟。如前所述执行洗涤步骤。通过在黑暗中与适当样品在冰上孵育30分钟,使用pe抗小鼠抗体(jir)检测vnar(his6myc标记)和相应vnar结合物的结合。如前所述执行洗涤步骤。将细胞颗粒重新悬浮在0.3ml冰冷pbs/2%fcs中,并在黑暗中置于冰上,之后在merck-millipore guava easycyte ht流式细胞仪进行分析。
[0324]
图16显示与vc-pab-eda-pnu或va-eda-pnu结合的vnar多聚体与癌细胞表面上的ror1保持结合。
[0325]
实施例5:组织蛋白酶b处理蛋白质-pnu结合物
[0326]
组织蛋白酶b处理的vnar-hfc-vc-pab-eda-pnu结合物和相应的多聚体蛋白结合物以定量方式释放出游离eda-pnu159682衍生物。然而,vnar-hfc-va-eda-pnu结合物和相应的多聚体蛋白结合物对组织蛋白酶b处理完全稳定。
[0327]
表11:用cat b处理后的有效载荷释放
[0328][0329]
在使用catb进行治疗时只有vc-pab-eda-pnu释放有效载荷,正如在这个体外实验中使用的条件所预期的。释放是定量的-由于结合物的药物抗体比为2,因此在catb治疗后,预计释放的有效载荷浓度是结合物浓度的两倍。该期望值与表11中vc-pab-eda-pnu结合物所示的数据相匹配。
[0330]
实施例6:蛋白质-pnu结合物的血浆稳定性
[0331]
不同的p3a1-hfc-pnu结合物在37℃以4μm蛋白质的最终浓度在小鼠血浆中孵育。通过lc-ms分析样品随时间的变化,并参考校准标准对释放的pnu159682和eda-pnu159682衍生物的量进行量化。
[0332]
表12:蛋白质-pnu结合物在小鼠血浆中孵育后的稳定性lc-ms检测到的游离pnu有效载荷浓度(nm)
[0333][0334]
如表12所示,具有va-eda-pnu159682有效载荷的结合物显示出优异的小鼠血浆稳定性,随着时间的推移,pnu衍生物几乎不释放。对于vc-pab-dmae-pnu和vc-pab-eda-pnu结合物二则,检测到的一些游离的有效载荷的释放随时间变化,120小时后分别检测到217nm pnu159682和114nm eda-pnu159682衍生物。注意,在平行研究中,发明人计算了游离pnu159682和eda-pnu159682衍生物的小鼠血浆半衰期分别为33小时和116小时。表明相对于vc-pab-eda-pnu结合物,vc-pab-dmae-pnu结合物释放的游离pnu有效载荷的绝对量被低
估。
[0335]
不同的结合物在37℃以4μm或2μm蛋白质的最终浓度在人血浆中孵育168小时。作为168小时(含168小时)内时间的函数,通过lc-ms对样品进行分析,并参考校准标准对释放的pnu159682和eda-pnu159682衍生物的量进行量化。
[0336]
表13:蛋白质-pnu结合物在人血浆中孵育后的稳定性
[0337][0338]
如表13所示,结合物显示出极好的人血浆稳定性,在实验过程中检测不到释放的pnu衍生物的量。eda-pnu在人血浆中的半衰期为172.25小时(4个不同实验的平均值)。
[0339]
实施例7:蛋白质-药物结合物在胸膜间皮瘤患者源性异种移植模型中的体内疗效
[0340]
查尔斯河实验室(弗莱堡)对ror1 pxf-1118患者源性胸膜间皮瘤异种移植模型进行了疗效研究。
[0341]
从裸鼠体内连续传代的异种移植物获得的肿瘤片段被皮下植入雌性nmri nu/nu小鼠(crl:nmri-foxn1
nu
)体内。对小鼠进行监测,直到在足够数量的动物中肿瘤植入物达到50mm3至250mm3,优选150mm3至200mm3的研究体积征集标准。将小鼠随机分配到治疗组,以使各组肿瘤体积之间无统计学差异。随机分组被指定为实验的第0天。在第1、4、7、10、18天,通过静脉注射0.3mg/kg的载体或蛋白质-药物结合物b1-hfc-vc-pab-eda-pnu或b1-hfc-va-eda-pnu治疗小鼠。所有小鼠在第一次pdc剂量前20小时以29mg/kg静脉注射(i.v.)小鼠igg接受单剂量启动。
[0342]
绝对肿瘤体积(atv)在随机化当天用数字卡尺进行二维测量,然后每周三次。根据
公式计算肿瘤体积:
[0343]
肿瘤体积=(l
×
w2)
×
0.5
[0344]
其中,l=肿瘤的最大直径,w=肿瘤的宽度(垂直直径)(单位:mm)。
[0345]
动物每周进行三次常规称重,并在给药当天进行称重。根据当地和最佳兽医实践指南,每天观察小鼠并记录其外观、行为、不良临床症状和一般健康的变化。
[0346]
图17显示了相对于载体对照蛋白质-药物结合物对肿瘤生长的影响。b1-hfc-vc-pab-eda-pnu和b1-hfc-va-eda-pnu二者均具有良好的耐受性并在ror1 胸膜间皮瘤pdx模型中显著抑制肿瘤生长。针对ror1 ve胸膜间皮瘤患者源性肿瘤的pdc分子显示出良好的抗肿瘤效果。
[0347]
实施例8:蛋白质-药物结合物在三阴性乳腺癌(tnbg)患者源性异种移植模型中的体内疗效
[0348]
xentech(巴黎)对ror1 hbcx-28患者源性tnbc异种移植模型进行了疗效研究。
[0349]
远交无胸腺(nu/nu)雌性小鼠(hsd:无胸腺裸鼠-foxn1
nu
)皮下移植相同体内传代的肿瘤。对小鼠进行监测,直到在足够数量的动物中肿瘤植入物达到60mm3至200mm3,优选75mm3至196mm3的研究体积征集标准。将小鼠随机分配到治疗组,以使各组肿瘤体积之间无统计学差异。随机分组被指定为实验的第0天。在第2、5、8、12、15天,通过静脉注射0.3mg/kg的载体或蛋白质-药物结合物b1-hfc-vc-pab-eda-pnu或b1-hfc-va-eda-pnu治疗小鼠,所有小鼠在第一次pdc剂量前20小时以小鼠igg预激发。在d55之前的实验期间,通过使用卡尺每周测量三次垂直肿瘤直径,然后每周测量和称重两次直到实验结束(即第103天)来评估肿瘤体积。使用公式tv(mm3)=[长度(mm)x宽度(mm)2]x 0.5计算绝对肿瘤体积(atv),其中长度和宽度分别为垂直测量的肿瘤的最长和最短垂直直径。所有动物在测量肿瘤大小的同时称重。根据当地福利和最佳兽医实践指南,每天观察小鼠并记录其外观、行为、不良临床症状和一般健康的变化。
[0350]
图18显示了相对于载体对照蛋白质-药物结合物对肿瘤生长的影响。b1-hfc-vc-pab-eda-pnu和b1-hfc-va-eda-pnu二者均具有良好的耐受性并在该ror1 tnbc pdx模型中显示出高度统计显著的体内疗效。另外,两种药物均观察到完全和持久的消退,包括在整个研究期间(103天)持续的肿瘤“治愈”。靶向ror1 ve tnbc患者源性肿瘤的pdc分子显示出强大的抗肿瘤疗效,肿瘤完全和持久的消退(包括“治愈”)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。