1.本发明属于医药化学技术领域,具体涉及一种小檗碱卡格列净衍生物及其制备方法和应用。
背景技术:
2.对天然药物进行结构改造和修饰,是药物研究者们开发新药的重要途径。小檗碱(berberine,bbr,c20h19no5,mw 336.37),又称黄连素,是从黄连(coptis chinensis)等中草药中分离提取得到的一种异喹啉类生物碱。是存在于黄连、黄柏、三颗针等传统中药中的活性生物碱,近年研究发现,其具有抗炎、降血糖、降血脂、抗病原微生物、抗肿瘤等多种药理活性,然需要高剂量才能达到一个好的降糖效果。小檗碱因具有独特的结构而在化学科学和医药的合成中广受欢迎。已有一些由小檗碱为原料实现通用分子向杂环化合物转化的报道。但由于小檗碱的脂溶性差,难以透过细菌的细胞膜,口服较难吸收,限制了其临床应用范围。因此通过分子结构改造来改变药物分子的结构特性并进而改变其体内外抑菌活性的相关研究则迫切需要。卡格列净(canagliflozin)是一种新型糖尿病治疗药物,是一种钠-葡萄糖共转运蛋白2(sglt2)抑制剂,但使用时也会有较多副作用,如急性肾损伤、女性外阴性霉菌感染等。卡格列净作为近年上市的新药,鲜有其结构修饰的报道。
技术实现要素:
3.有鉴于此,本发明的目的在于提供一种具有更强抑菌活性的小檗碱卡格列净衍生物,还提供该衍生物的制备方法和应用。
4.为达到上述目的,本发明提供如下技术方案:
5.1.一种小檗碱卡格列净衍生物,其结构式如式i所示:
[0006][0007]
2.一种小檗碱卡格列净衍生物的制备方法,具体包括以下步骤:
[0008]
a.将盐酸小檗碱还原得到小檗碱;
[0009]
b.对卡格列净的羟基硅醚先溴化得到产物11,再乙酸化得到产物4;
[0010][0011]
c.最后将小檗碱和产物11或者小檗碱和产物4反应得到结构式如式i所示的小檗碱卡格列净衍生物。
[0012]
进一步,所述的小檗碱卡格列净衍生物的制备方法,步骤a中将盐酸小檗碱还原得到小檗碱具体步骤为:取盐酸小檗碱溶于缚酸剂中,加入1-4eq的nabh4,0℃-25℃反应0.5-3h后,通过水洗得到产物还原小檗碱。
[0013]
进一步,所述的小檗碱卡格列净衍生物的制备方法,步骤b中产物11制备具体步骤为:将卡格列净溶于dmf中,开始搅拌,氩气保护条件下,加入三苯基膦,反应液降温至0℃,向反应体系中缓慢加入nbs,滴加完毕后,在50℃条件下反应4小时,通过柱层析分离得到产物11。
[0014]
进一步,所述的小檗碱卡格列净衍生物的制备方法,步骤b中产物11制备具体步骤中,柱层析的淋洗剂为二氯甲烷:甲醇20:1-40:1。
[0015]
进一步,所述的小檗碱卡格列净衍生物的制备方法,将产物11溶于吡啶,加入乙酸酐,室温条件下搅拌反应4小时,再通过柱层析分离得产物4。
[0016]
进一步,所述的小檗碱卡格列净衍生物的制备方法,柱层析分离得产物4的淋洗剂为石油醚:乙酸乙酯10:1。
[0017]
进一步,所述的小檗碱卡格列净衍生物的制备方法,步骤c制备具体步骤为:取产物4或者产物11和还原小檗碱于无水乙腈中,加入碘化合物,于80℃-130℃条件下反应10-24小时,通过柱层析分离得到目标产物,为橙黄色固体。
[0018]
进一步,所述的小檗碱卡格列净衍生物的制备方法,所述碘化合物为nai。
[0019]
进一步,所述的小檗碱卡格列净衍生物的制备方法,步骤c反应在氩气保护的条件下进行。
[0020]
3.上述任一项技术方案所述的小檗碱卡格列净衍生物在制备抗细菌药物中的应用。
[0021]
进一步,所述的小檗碱卡格列净衍生物在制备抗细菌药物中的应用,所述细菌为革兰氏阳性菌和革兰氏阴性菌。
[0022]
进一步,所述的小檗碱卡格列净衍生物在制备抗细菌药物中的应用,所述细菌为大肠杆菌、金黄色葡萄球菌和/或铜绿假单胞菌。
[0023]
本发明的有益效果在于:本发明通过多次试验,在不改变小檗碱与卡格列净基本骨架的条件下,将盐酸小檗碱(bbr)与卡格列净(can)通过化学合成的方法链接,合成一种全新的化合物bc,具有一定的降糖效果,展现出优异的抗菌活性和广谱抗菌作用,而且可同时抑制革兰氏阳性菌和革兰氏阴性菌的活性,同bbr和can相比,mic值大大降低了,尤其是对于铜绿假单胞菌,大大减少了患者的用药量,有利于防止耐药性微生物菌株的产生。本发明还涉及化合物的制备方法,产率高,副反应少,产品纯度高,反应设备要求低,易于工业化
生产。
附图说明
[0024]
为了使本发明的目的、技术方案和有益效果更加清楚,本发明提供如下附图进行说明:
[0025]
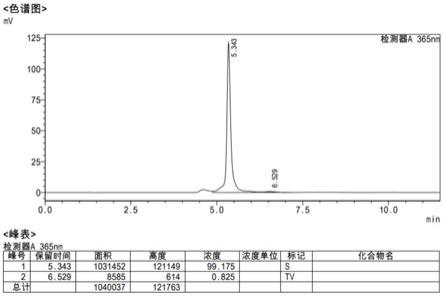
图1为产物bc的高效液相色谱图;
[0026]
图2为各药物组的降糖效果示意图;
[0027]
图3为各药物组的体外炎症因子抑制实验结果图;
[0028]
图4为各药物组的抗菌实验结果图;
[0029]
图5为can、bbr、bbr can、bc在0.1mm浓度下24h对细菌生物膜形成的抑制作用示意图;
[0030]
图6为用0.1mm的化合物bc处理24小时的铜绿假单胞菌胞内可溶性蛋白的sds-page分析。
[0031]
图7—图10依次为化合物bc的1h、
13
c、
19
f-nmr和高分辩质谱(hrms)谱图;
[0032]
图11-图13依次为化合物ba-c的1h、
13
c、
19
f-nmr谱图。
[0033]
图14为酯化卡格列净的1h
–
nmr。
[0034]
图15-图17依次为溴化卡格列净(br-c)的1h、
13
c、
19
f-nmr。
[0035]
图18为还原小檗碱1h nmr图。
具体实施方式
[0036]
下面将结合附图,对本发明的优选实施例进行详细的描述。实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。
[0037]
本发明通过多次试验,最后在不改变原料化合物基本骨架的条件下,将盐酸小檗碱(bbr)与卡格列净(can)通过化学合成的方法链接,合成一种全新的具有药物应用前景的化合物bc。原料及目标产物结构式如下:
[0038][0039]
目标产物bc:
[0040][0041]
实施例1
[0042]
设计了如下路线(路线1):
[0043]
反应1:对盐酸小檗碱1进行还原得到还原小檗碱(hb);将盐酸小檗碱(1g,2.66mol)溶于15ml吡啶中后,加入2eq的nabh4,室温反应1h后,通过水洗可得到产物2还原小檗碱,产率为65%。
[0044][0045]
在上述还原小檗碱的制备方法中:取盐酸小檗碱(1g,2.66mol)溶于15ml吡啶(其他缚酸剂亦可)中,加入1-4eq的nabh4,0℃-25℃反应0.5-3h后,通过水洗都可得到产物还原小檗碱2。
[0046]
该反应方法还有,将盐酸小檗碱溶于含碳酸钾的甲醇溶液中,0℃也可以进行反应,但需反应8小时,产率为46%。
[0047]
反应2(反应步骤a):对卡格列净进行酯化得到产物3(ac-c);将卡格列净溶于吡啶中,加入乙酸酐,反应4h,得到产物3。
[0048]
反应3(反应步骤b):对产物3进行溴化得到产物4(ba-c);但是反应3未能检测到反应进行,即未能得到预期的产物。在该反应中,不论是改变溶剂、改变反应体系配比还是改变反应体系温度,都未能检测到反应进行。以至后续的反应未能顺利进行。卡格列净衍生物的合成:试剂和条件:a)乙酸酐,吡啶,rt,4h,82%b)pbr3,ch2cl2,0℃,12h。
[0049][0050]
产物3(2r,3r,4r,5s,6r)-2-(乙酰氧基甲基)-6-(3-(5-(4-氟苯基)噻吩-2-基)甲基)-4-甲基苯基)四氢-2h-吡喃-3,4,5-三乙酸酯的制备:取卡格列净(1mmol)溶于1ml吡啶,加入乙酸酐(5mmol),室温条件下搅拌反应4小时,再通过柱层析得白色固体(82%),淋洗剂为石油醚:乙酸乙酯10:1(体积比)。
[0051]1h nmr(400mhz,cdcl3)δ7.51
–
7.43(m,2h),7.18(d,j=7.1hz,3h),7.01(d,j=8.8hz,3h),6.61(s,1h),5.32(t,j=9.2hz,1h),5.23(t,j=9.5hz,1h),5.14(t,j=9.4hz,1h),4.36(d,j=9.7hz,1h),4.33
–
4.25(m,1h),4.19
–
4.04(m,3h),3.82(d,j=7.6hz,1h),2.29(s,3h),2.06(d,j=3.0hz,6h),2.00(s,3h),1.76(s,3h).)
[0052]
反应4(反应步骤c):将还原小檗碱2与产物4相连得到产物5;
[0053]
反应5(反应步骤d):对产物5进行水解得到终产物——产物6。目的在于将卡格列净羟基硅醚处的羟基转为卤素基团取代,增加与还原小檗碱反应的活性,使其在结构上易与小檗碱相连。
[0054][0055]
实施例2
[0056]
为得到产物4(ba-c),继续尝试通过羟基硅醚保护与脱保护的方法,对卡格列净进行修饰,做了以下尝试(路线2):
[0057]
反应6(反应步骤e):对卡格列净羟基硅醚处的羟基进行保护得到产物7;
[0058]
反应7(反应步骤f):对产物7进行酯化得到产物8;
[0059]
反应8(反应步骤g):对产物8进行脱保护得到产物9;
[0060]
反应9(反应步骤h):对产物9进行溴化得到产物10。
[0061][0062]
值得一提的是,反应6中我们尝试用了tbdpscl(叔丁基二苯基氯硅烷)、三甲基氯硅烷、tipscl(三异丙基氯硅烷)作为保护基,通过对产物体系进行tlc点板发现只有使用了tbdpscl作为保护基时才可检测到目标物质。反应7成功检测到目标产物,且产率很高。然反应8未能检测到目标产物,以至后续反应未能顺利进行。
[0063]
实施例3
[0064]
经过多次尝试,最后使用直接对卡格列净进行先溴化后酯化的合成(路线3):
[0065]
反应10(反应步骤i):卡格列净衍生物的合成,试剂及条件:i)cbr4,pph3,py,ar,50℃
[0066][0067]
反应10:取卡格列净(1mmol)溶于2ml超干吡啶,氩气保护条件下,开始搅拌,加入三苯基膦(2mmol)。反应液降温至0℃,向反应体系中缓慢加入四溴化碳(1mmol),滴加完毕后,在50℃条件下反应4小时,通过柱层析分离得到产物,展开剂为二氯甲烷:甲醇40:1,但通过核磁进行初步表征,非目标产物。
[0068]
反应11(反应步骤j和反应步骤k):卡格列净衍生物的合成,试剂及条件:j)nbs,
pph3,dmf,ar,50℃,4h;k)乙酸酐,吡啶,rt,4h
[0069][0070]
产物11的制备:将卡格列净(1mmol)溶于1mldmf中,开始搅拌,氩气保护条件下,加入三苯基膦(2mmol)。反应液降温至0℃,向反应体系中缓慢加入nbs(2mmol),滴加完毕后,在50℃条件下反应4小时,通过柱层析分离得到产物11,为白色固体粉末(72%)。(柱层析填料可为200-300目的层析用硅胶、三氧化二铝,硅藻土等常用填料),淋洗剂为二氯甲烷:甲醇20:1-40:1(体积比)。
[0071]
产物4ba-c的制备:将上步反应得到的白色固体(1mmol)溶于1ml吡啶,加入乙酸酐(5mmol),室温条件下搅拌反应4小时,再通过柱层析得白色固体(80%),淋洗剂为石油醚:乙酸乙酯10:1(体积比)。
[0072]1h nmr(400mhz,cdcl3)δ7.47(dd,j=8.7,5.3hz,2h),7.18(q,j=7.6hz,3h),7.06
–
6.97(m,3h),6.62(d,j=3.5hz,1h),5.32(t,j=9.4hz,1h),5.21(t,j=9.5hz,1h),5.12(t,j=9.6hz,1h),4.40(d,j=9.8hz,1h),4.17
–
4.03(m,2h),3.86
–
3.78(m,1h),3.54(dd,j=11.4,2.7hz,1h),3.43(dd,j=11.4,5.5hz,1h),2.29(s,3h),2.08(s,3h),2.00(s,3h),1.76(s,3h).
[0073]
13
c nmr(100mhz,cdcl3)δ170.42,169.46,168.82,163.30,160.84,143.07,141.56,138.11,137.09,134.23,130.78,128.39,127.13,127.05,126.04,125.46,122.72,115.84,115.63,79.68,76.74,74.13,72.75,71.00,33.98,31.46,31.22,30.22,29.71,20.72,20.68,20.43,19.31.
[0074]
19
f nmr(300mhz,cdcl3)δ-115.08.)
[0075]
反应10的产物体系通过tcl荧光检测有新化合物生成,但通过h谱表征后发现所得物质非目标化合物,后使用nbs进行j反应,终于得到了目标化合物11溴化卡格列净(br-c)和产物4(ba-c)。
[0076]
得到目标产物4后进行路线1中的反应4和反应5,同时,直接尝试br-c与小檗碱的反应,得到如下路线(路线4,反应步骤i),为目标产物的最终合成路线。bc的合成,试剂及条件:l)nai,mecn,ar,130℃16h。
[0077]
bc的制备方法1:取br-c(1mmol)和还原小檗碱hb(1.5mmol)于2ml无水乙腈中,在氩气保护的条件下,加入nai(0.1mmol,0.1-0.5mmol都可以),于130℃条件下反应16小时,通过柱层析(用硅胶、三氧化二铝,硅藻土等常用填料),淋洗剂为按体积比计的二氯甲烷:
甲醇:三乙胺=20:1:0.2%-1:1:0.2%分离得到目标产物,为橙黄色固体(60%)。(80℃也可以反应,产率为40%,无氩气保护条件下产率为26%)。也可以使用其他相似含碘化合物、或通过调节反应温度和时间达到相同的效果。
[0078]
bc的制备方法2:取ba-c(1mmol)和还原小檗碱hb(1.5mmol)于2ml无水乙腈中,在氩气保护的条件下,加入nai(0.1mmol,0.1-0.5mmol都可以),于130℃条件下反应16小时,通过柱层析(用硅胶、三氧化二铝,硅藻土等常用填料),淋洗剂为按体积比计的二氯甲烷:甲醇:三乙胺=20:1:0.2%-1:1:0.2%分离得到目标产物,为橙黄色固体。
[0079]1h nmr(400mhz,cd3od)δ9.83(s,1h),8.88(s,1h),8.15(d,j=9.1hz,1h),7.95(d,j=9.0hz,1h),7.74(s,1h),7.64(d,j=16.1hz,2h),7.54(dd,j=7.9,5.2hz,1h),7.38
–
7.29(m,1h),7.21(t,j=7.7hz,1h),7.17
–
7.07(m,2h),7.04(s,1h),6.12(s,2h),5.70(s,1h),5.02
–
4.78(m,3h),4.17(t,j=6.5hz,3h),4.04(d,j=10.8hz,9h),3.18
–
3.12(m,3h),2.23(t,j=12.0hz,3h),2.13(t,j=7.4hz,2h),1.63
–
1.55(m,3h).
[0080]
13c nmr(100mhz,cd3od)δ177.82,172.99,152.12,151.96,149.87,145.71,139.59,135.09,134.94,134.84,134.45,134.34,131.84,131.50,131.37,131.13,131.00,130.94,128.26,128.07,126.11,124.54,123.26,121.80,121.47,109.39,106.53,103.65,62.59,57.66,57.21,36.63,35.37,35.27,33.04,30.91,30.75,30.45,26.73,23.71,22.59,22.55,14.71,14.44.
[0081]
19f nmr(300mhz,cd3od)δ-115.39.)
[0082][0083]
需要注意的是,该反应在无氩气保护和130℃高温条件下均可反应,但产率偏低。
[0084]
bc的纯度测定:高效液相色谱法,用ods c18色谱柱,以乙腈:h3po4水溶液(ph=3)v:v=98:2为流动相,在365nm波长处,以0.5ml/min的流速测定了产物的纯度为99.175%。图1为产物bc的高效液相色谱图。
[0085]
实施例4
[0086]
为评价bbr-can化合物(bc)相较于bbr和can药物联用的降糖效果。我们利用链脲佐菌素诱导的糖尿病雄性nih小鼠,动物分组后,通过灌胃的方式给予药物小檗碱(bbr)、卡格列净(can)、bbr-can化合物(简写bc)、bbr can联合用药(简写b c),以及溶解药物所用溶剂0.5%cmc-na作为对照,每天给药1次,在给药第6天测定禁食6h血糖。最终结果如图2所示:bc的降糖效果优于bbr,但没有达到bbr与can联合使用的效果。推测,有可能改变卡格列
净羟基硅醚处的羟基后,影响了其原本对钠-葡萄糖共转运蛋白2(sglt2)的抑制效果。
[0087]
为评价bbr-can化合物相较于bbr和can药物联用的抗炎效果。我们在脂多糖诱导的小鼠单核巨噬细胞j774a.1上进行了体外炎症因子抑制实验并得到了以下结果,如图3所示,由图可以看出,bc对il-1β和tnf-α等炎症因子的抑制效果都优于bbr、can、bbr can联合用药。
[0088]
为进一步评价bbr-can化合物相较于bbr和can药物联用的抗炎效果。我们根据美国临床实验室标准化协会的相关标准,采用微量肉汤稀释法检测不同药物的mic值,最终发现bbr、b c和bc对金黄色葡萄球菌、铜绿假单胞菌和大肠杆菌均表现出抗菌活性,复方bc显示出较强的抗菌活性和广谱抗菌作用,大大优于bbr、b c,可同时抑制革兰氏阳性菌和革兰氏阴性菌的活性。b c的抑菌效果优于单独使用bbr,说明bbr和can可产生一定的协同抑菌效果。复方bc抗菌作用最强,优于bbr与can的协同作用。结果如下表1和图4所示。
[0089]
表1 bbr、bbr can、bc对不同菌株的最低抑菌浓度(mics)
[0090][0091][0092]
图4为can、bbr、bbr can、bc对大肠杆菌、金黄色葡萄球菌和铜绿假单胞菌生长的影响,曲线从上到下依次对应右边的图例标记,其中ck代表对照组,与bc组相比*p《0.05,**p《0.01,***p《0.001。(a)-(c)为铜绿假单胞菌、金黄色葡萄球菌,大肠杆菌对can、bbr、bbr can、bc在mic下的抑制作用随时间的变化。(d)-(f)为铜绿假单胞菌、金黄色葡萄球菌,大肠杆菌对can、bbr、bbr can、bc在1/2mic下的抑制作用随时间的变化。
[0093]
图5为can、bbr、bbr can、bc在0.1mm浓度下24h对细菌生物膜形成的抑制作用,使用0.1%的结晶紫进行染色观察。其中细菌菌株:(a)铜绿假单胞菌、(b)金黄色葡萄球菌、(c)大肠杆菌,(d)为铜绿假单胞菌的结晶紫染色图。bbr、b c、bc对铜绿假单胞菌的生物膜形成有抑制作用,其中bc的抑制作用最为明显;对于大肠杆菌和金黄色葡萄球菌,can对生物膜形成的影响不大,bbr对生物膜形成的抑制作用较弱,但b c和bc组对生物膜形成的抑制作用明显,bc组抑制效果最好。图5d显示了结晶紫染色吸附在孔壁上的生物膜的结果,可以更直观地看出,bc的抑制作用最强。
[0094]
图6为0.1mm bc处理24h后铜绿假单胞菌胞内可溶性蛋白的sds-page分析。bc处理
的铜绿假单胞菌胞内可溶性蛋白与对照组有显着差异。bc处理后的蛋白条带明显变浅、模糊,说明bc对铜绿假单胞菌胞内蛋白具有破坏作用,从而达到抑菌的效果。bbr和b c组的蛋白条带也较对照组有所减弱,但效果远低于bc处理组。
[0095]
图7—图9依次为化合物bc的1h、
13
c、
19
f-nmr;图10为化合物bc的高分辩质谱(hrms)谱图;图11-图13依次为化合物ba-c的1h、
13
c、
19
f-nmr谱图;图14为酯化卡格列净的1h
–
nmr;图15-图17依次为溴化卡格列净(br-c)的1h、
13
c、
19
f-nmr;图18为还原小檗碱1h nmr图。
[0096]
最后说明的是,以上优选实施例仅用以说明本发明的技术方案而非限制,尽管通过上述优选实施例已经对本发明进行了详细的描述,但本领域技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离本发明权利要求书所限定的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。