1.本发明涉及医药技术领域,尤其是涉及新型糖苷类化合物、药物组合物、制备方法及应用。
背景技术:
2.随着人民生活及工作方式的日益改变,糖尿病的发病率呈现逐年上升趋势。糖尿病病程的持续发展可能会引起一系列并发症,进而影响患者的生活质量并加重患者及国家的经济负担。因此,具有预防或者治疗糖尿病的药物的研发受到越来越多的关注。
3.狼毒大戟(euphorbia fischeriana steud.)为大戟科大戟属多年生草本植物,关于该植物的研究多集中在萜类化合物及抗肿瘤活性上。例如专利cn104622865a公开了巨大戟二萜类化合物在制备抗肿瘤药物中的应用,专利cn103623033a公开了用于抑制癌症及/或肿瘤细胞生长的大戟科植物活性物质及其应用,这些专利均显示大戟科植物的活性提取物质对于抗癌或者抗肿瘤(例如促进肿瘤细胞死亡)的重要作用。
4.目前几乎没有或很少有关于糖苷类化合物或大戟科植物提取物在糖尿病方面发挥作用的相关研究和报道。
5.鉴于此,特提出本发明。
技术实现要素:
6.本发明的目的在于提供新型糖苷类化合物、药物组合物、制备方法及应用,以丰富现有技术中糖尿病相关药物的类型,为糖尿病及其相关病症提供新的治疗途径。
7.为了解决上述技术问题,本发明提供的技术方案如下:
8.在一个实施方案中,本发明提供了一类新型糖苷类化合物,其具有式(i)的母核结构:
[0009][0010]
其中,r1和r2各自分别选自氢、酯基、甲基、乙基、丙基、异丙基、没食子酰基、氨基、甲氨基、二甲氨基、乙胺基、二乙胺基、异丙氨基、二异丙氨基、饱和的烃基、不饱和的烃基、取代的芳香环、未取代的芳香环、低聚糖、多聚糖、金属离子、有机酸酯基、硝基或卤素中的任一种。
[0011]
本发明从狼毒大戟中寻找高效低毒的生物活性成分,从该植物中分离得到一类糖
苷类化合物,该类化合物具有显著的α-葡萄糖苷酶抑制活性。
[0012]
在一个实施方案中,所述糖苷类化合物中,
[0013]
r1为r2为该化合物命名为euphorbiacetophenone e。
[0014]
本发明化合物euphorbiacetophenone e的其分子结构式如下:
[0015][0016]
在一个实施方案中,所述糖苷类化合物中,
[0017]
r1为r2为该化合物命名为1,2,3-tri-o-galloyl-β-d-glucopyranose。
[0018]
本发明化合物1,2,3-tri-o-galloyl-β-d-glucopyranose的其分子结构式如下:
[0019][0020]
在另一个方面,本发明提供了一种药物组合物,该药物化合物含有前述新型糖苷类化合物。
[0021]
在一个实施方案中,所述药物组合物包含联用药物;优选地,所述联用药物包括以下的一种或多种:胰岛素、甲苯磺丁脲、氯磺丙脲、格列苯脲、格列吡嗪、格列齐特、格列美脲、格列波脲、格列喹酮、二甲双胍、盐酸二甲双胍、苯乙双胍、吡格列酮、盐酸吡格列酮、罗格列酮、马来酸罗格列酮、曲格列酮、环格列酮、恩格列酮、阿格列汀、苯甲酸阿格列汀、沙格列汀、西格列汀、磷酸西格列汀、维格列汀、利格列汀、糖-100、阿卡波糖、伏格列波糖、米格列醇、瑞格列奈、那格列奈、米格列奈、依克那肽、艾塞那肽、利拉鲁肽、普林兰肽、醋酸普兰
林肽;
[0022]
更优选地,所述胰岛素包括低精蛋白锌胰岛素、珠蛋白锌胰岛素或精蛋白锌胰岛素中的一种或多种。
[0023]
在一个实施方案中,所述药物组合物还包含药学上可接受的载体或赋形剂;“药学上可接受的载体”指用于治疗剂给药的载体,包括各种赋形剂和稀释剂。通常,载体并不是必要的活性成分,且施用后没有过分的毒性。在组合物中药学上可接受的载体可包括液体,如水、盐水、甘油和乙醇。在一些情况系啊,赋形剂包括填充剂、粘合剂、润滑剂、崩解剂、助溶剂、表面活性剂等类型。合适的载体和或赋形剂是本领域普通技术人员所熟知的,可以根据实际情况进行选择。可将本发明的药物活性成分与一种或多种固体或液体药物赋形剂和/或辅剂结合,制成可作为人药或兽药使用的适当的施用形式或剂量形式。
[0024]
在本发明中,所述药物组合物的剂型包括片剂、胶囊剂、颗粒剂、口服液、糖浆剂、膏剂、冲剂、滴丸或微丸中的任一种。例如片剂包括但不限于普通片、肠溶片、含片、分散片、咀嚼片、泡腾片、口腔崩解片;胶囊剂包括但不限于硬胶囊、软胶囊、肠溶胶囊;注射剂包括但不限于水针剂、粉针剂和输液剂。本发明的药物还可以制成本领域常见的其他剂型,例如溶液剂包括但不限于真溶液和胶体溶液;乳剂包括但不限于o/w型、w/o型和复乳;半固体剂型包括但不限于软膏剂、凝胶剂、糊剂等;混悬剂、散剂、栓剂、膜剂、贴片、气(粉)雾剂、喷雾剂等。
[0025]
在一个实施方案中,药物组合物中含有有效量的所述糖苷类化合物;例如在所述药物组合物中,所述糖苷类化合物的重量占比为0.01至95%,包括但不限于0.01、0.1、0.2、0.5、1、1.5、2、3、3、4、5、6、7、8、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90和95%。
[0026]
在一个优选的方案中,所述药物组合物的单元剂型中所述糖苷类化合物的重量占比为0.01至50mg,包括但不限于1、5、10、15、20、25、30、35、40、45、50mg;更为优选地,重量占比为0.01至10mg。
[0027]
在另一个方面,本发明提供了所述糖苷类化合物的制备方法,所述糖苷类化合物是从狼毒大戟(euphorbia fischeriana steud.)中分离获得;
[0028]
进一步地,将原料狼毒大戟根部干燥粉碎后用溶剂提取,收集提取液并浓缩得到浓缩液;用有机溶剂对所述浓缩液进行萃取获得萃取物;将所述萃取物经硅胶柱层析,洗脱后的得到的第一目的流分进行反相硅胶柱色谱层析;将反相硅胶柱色谱层析得到的第二目的流分进行葡聚糖凝胶柱层析,然后,通过反相或正相制备液相色谱纯化,得到化合物。
[0029]
在一个实施方案中,所述制备方法具体包括:
[0030]
(a)取狼毒大戟根部,干燥并粉碎,利用提取溶剂提取,合并提取液,过滤,减压浓缩,得浓缩液;
[0031]
(b)使用有机溶剂对所述浓缩液进行萃取,合并有机相,获得萃取物;
[0032]
(c)对所述萃取物进行硅胶柱层析,使用洗脱剂进行梯度洗脱,获取第一目的留分进行反相硅胶柱色谱层析洗脱获取第二目的流分;将所述第二目的流分进行葡聚糖凝胶柱层析并洗脱;
[0033]
(d)通过反相或正相制备液相色谱纯化,得到化合物。
[0034]
在一个实施方案中,步骤(a)中,所述提取溶剂的质量为所述狼毒大戟根部原料质
量8~15倍,优选10倍;优选地,所述提取为回流提取;
[0035]
进一步地,所述回流提取的时间为1~3小时,温度为65℃~85℃,回流提取的次数为2~4次;更优选地,所述提取溶剂选自水、甲醇或者乙醇中的一种或多种;最优选地,提取溶剂为60%~95%乙醇;
[0036]
步骤(b)中,用水分散后,使用有机溶剂萃取2~5次;优选地,所述有机溶剂包括二氯甲烷、三氯甲烷、环乙烷、乙酸乙酯、乙酸丙酯和石油醚中的一种或多种,优选乙酸乙酯、乙酸丙酯;优选地,所述有机溶剂的用量为所述浓缩液体积的1.5~2倍;
[0037]
步骤(c)中,所述梯度洗脱的洗脱剂包括氯仿-甲醇、石油醚-丙酮、石油醚-乙酸乙酯、环己烷-丙酮或者环己烷-乙酸乙酯中的任一种;优选石油醚-乙酸乙酯、环己烷-乙酸乙酯;所述反相硅胶柱色谱层析的洗脱剂选自乙醇-水或甲醇-水中的任一种;所述葡聚糖凝胶柱层析的洗脱剂选自甲醇、氯仿-甲醇、二氯甲烷-甲醇、石油醚-氯仿-甲醇,中的任一种,优选甲醇、二氯甲烷-甲醇;优选地,所述葡聚糖凝胶选自sephadex lh-20,sephadex g-10、g-15、g-25、g-50、g-75、g-100、g-150和g-200,优选sephadex lh-20;
[0038]
步骤(d)中,色谱纯化处理中的色谱柱选自c4、c8、c6或c18中的任一种;其中,优选c18;流动相选自甲醇-水或乙腈-水中的任一种。
[0039]
在一个具体的实施方案中,化合物euphorbiacetophenone e和1,2,3-tri-o-galloyl-β-d-glucopyranose的制备方法,包括如下步骤:
[0040]
步骤1:取狼毒大戟根部,干燥并粉碎,利用提取溶剂进行若干次回流提取,合并提取液,过滤,减压浓缩,得浓缩液;
[0041]
步骤2:使用有机溶剂将步骤s10中得到的浓缩液萃取若干次,合并有机相,回收溶剂,获得萃取物iii;
[0042]
步骤3:对萃取物iii进行硅胶柱层析,使用洗脱剂进行梯度洗脱,利用硅胶薄层板检识后,合并依次得到流分a、流分b、流分c、流分d和流分e;
[0043]
将流分d进行反相硅胶柱色谱层析,通过硅胶薄层板检识获得(6个流分d1-d6)流分d1、流分d2、流分d3、流分d4、流分d5和流分d6;对流分d3进行葡聚糖凝胶柱层析;
[0044]
步骤4:分别通过反相或正相制备液相色谱纯化,得到化合物euphorbiacetophenone e和1,2,3-tri-o-galloyl-β-d-glucopyranose。
[0045]
在另一个方面,本发明提供了所述糖苷类化合物或含有糖苷类化合物的药物组合物在制备预防或治疗糖尿病及其并发症的药物中的应用。
[0046]
本发明提供了所述糖苷类化合物在制备α-葡萄糖苷酶抑制剂类降糖药物制剂(例如α-葡萄糖苷酶抑制剂)中的应用。
[0047]
本发明以该类糖苷类化合物为原料,经α-葡萄糖苷酶抑制活性筛选试验,结果显示euphorbiacetophenone e和1,2,3-tri-o-galloyl-β-d-glucopyranose抑制α-葡萄糖苷酶活性均优于阳性药阿卡波糖(ic
50
=286.23
±
0.86μm)。其中,化合物1,2,3-tri-o-galloyl-β-d-glucopyranose抑制活性最显著,ic
50
值为15.48
±
0.19μm。对1,2,3-tri-o-galloyl-β-d-glucopyranose进行酶抑制动力学试验发现,该化合物为α-葡萄糖苷酶混合型抑制剂。
[0048]
有益效果:本发明提供发现了一类糖苷类化合物,经α-葡萄糖苷酶抑制活性筛选试验和抑制动力学试验表明,该类化合物对α-葡萄糖苷酶具有明显的抑制作用,可以作为
治疗糖尿病疾病中的药物进行应用。也可以用于制备降血糖的保健食品或功能食品方面。
附图说明
[0049]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0050]
图1为本发明1,2,3-tri-o-galloyl-β-d-glucopyranose对α-葡萄糖苷酶活性的抑制作用图;
[0051]
图2为本发明1,2,3-tri-o-galloyl-β-d-glucopyranose对α-葡萄糖苷酶活性的抑制动力学曲线图;
[0052]
图3为为本发明1,2,3-tri-o-galloyl-β-d-glucopyranose对α-葡萄糖苷酶活性的michaelis-menten图(米海利斯-曼恬图,也称米氏曲线);
[0053]
图4为本发明1,2,3-tri-o-galloyl-β-d-glucopyranose对对α-葡萄糖苷酶活性的lineweaver-burk图(即,双倒数作图)。
具体实施方式
[0054]
下面将结合实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0055]
实施例1.糖苷类化合物euphorbiacetophenone e和1,2,3-tri-o-galloyl-β-d-glucopyranose的制备方法
[0056]
步骤s1:取狼毒大戟根部,干燥并粉碎,加入8-15倍量的溶剂回流提取1-3小时,重复2-4次,合并提取液,过滤,减压浓缩,得浓缩液;提取温度为65℃-85℃;
[0057]
提取溶剂可选自水、甲醇、乙醇或其混合物,优选60-95%乙醇;(本实施例中加入10倍量的80%乙醇溶剂,回流提取2小时,重复3次)。
[0058]
步骤s2:将浓缩液通过有机溶剂萃取2-5次,合并有机相,回收有机溶剂,得萃取物iii;提取温度为65℃至提取溶剂的沸点温度;
[0059]
有机萃取溶剂可为二氯甲烷、三氯甲烷、环乙烷、乙酸乙酯、乙酸丙酯和石油醚,优选乙酸乙酯、乙酸丙酯;(本实施例中将浓缩液通过有机溶剂萃取4次,溶剂为乙酸乙酯)。
[0060]
步骤s3:将萃取物iii通过硅胶柱层析,采用有机试剂进行梯度洗脱,并利用硅胶薄层板检识,合并为5个流分(a-e);随后选择d进行反相硅胶柱色谱层析,通过硅胶薄层板检识共得6个流分(d1-d6);对d3进行葡聚糖凝胶柱层析;
[0061]
硅胶柱层析洗脱剂为石油醚-乙酸乙酯、石油醚-丙酮、环己烷-乙酸乙酯、环己烷-丙酮、氯仿-甲醇,优选石油醚-乙酸乙酯、环己烷-乙酸乙酯;反相硅胶柱色谱层析的洗脱剂可选择乙醇-水或甲醇-水;葡聚糖凝胶柱层析所用材料可选择sephadex lh-20,sephadex g-10,g-15,g-25,g-50,g-75,g-100,g-150和g-200;葡聚糖凝胶柱层析洗脱剂可选择甲醇、氯仿-甲醇、二氯甲烷-甲醇、石油醚-氯仿-甲醇,优选甲醇、二氯甲烷-甲醇(在本实施例中
硅胶柱层析洗脱剂为石油醚-乙酸乙酯,反相硅胶柱色谱层析的洗脱剂为乙醇-水,葡聚糖凝胶柱层析的材料为sephadex lh-20,洗脱剂为甲醇)。
[0062]
步骤s4:分别通过反相制备液相色谱纯化得到化合物euphorbiacetophenone e和1,2,3-tri-o-galloyl-β-d-glucopyranose;
[0063]
反相制备液相色谱柱可选择c4、c6、c8或c18,优选c18;流动相可选择甲醇-水或乙腈-水(本实施例中选择c18液相色谱柱,流动相选择甲醇-水)。
[0064]
实施例2
[0065]
其他与实施例1相同,不同之处在于:
[0066]
步骤s1:本实施例中加入8倍量的50%乙醇溶剂,回流提取1小时,重复2次。
[0067]
步骤s2:本实施例中将浓缩液通过有机溶剂萃取2次,溶剂为乙酸丙酯。
[0068]
步骤s3:在本实施例中硅胶柱层析洗脱剂为石油醚-丙酮,反相硅胶柱色谱层析的洗脱剂为甲醇-水,葡聚糖凝胶柱层析的材料为sephadex g-10,洗脱剂为二氯甲烷-甲醇。
[0069]
步骤s4:本实施例中选择c4液相色谱柱,流动相选择乙腈-水。
[0070]
实施例3
[0071]
其他与实施例1相同,不同之处在于:
[0072]
步骤s1:本实施例中加入15倍量的95%乙醇溶剂,回流提取3小时,重复4次。
[0073]
步骤s2:本实施例中将浓缩液通过有机溶剂萃取5次,溶剂为乙酸丙酯。
[0074]
步骤s3:在本实施例中硅胶柱层析洗脱剂为环己烷-乙酸乙酯,反相硅胶柱色谱层析的洗脱剂为甲醇-水,葡聚糖凝胶柱层析的材料为sephadex g-10,洗脱剂为石油醚-氯仿-甲醇。
[0075]
步骤s4:本实施例中选择c6液相色谱柱,流动相选择乙腈-水。
[0076]
需要说明的是,本发明实施例2-3以及本发明未示出的其他制备方法实例均与本发明实施例1制备结果类似;只要在本发明的制备方法所限定的参数条件下均能准备本发明的糖苷类化合物euphorbiacetophenone e和1,2,3-tri-o-galloyl-β-d-glucopyranose。
[0077]
实施例4
[0078]
本发明化合物euphorbiacetophenone e的其分子结构式如下:
[0079][0080]
euphorbiacetophenone e的物理性质为白色无定型粉末;核磁数据如下:
[0081]
uv(meoh)λ
max 215,280nm;ir(kbr)ν
max 3396,1715,1619cm
–1;hr-esi-ms:m/z 647.1254[m-h]-(cal.647.1254);1h nmr(500mhz,dmso-d6):δ
h 5.72(1h,d,j=8.0hz,h-1),5.18(1h,dd,j=9.8,8.0hz,h-2),5.42(1h,t,j=9.5hz,h-3),3.65(1h,t,j=9.5hz,h-4),3.81(1h,overlap,h-5),3.42(1h,dd,j=12.0,3.0hz,h-6α),3.33(1h,dd,j=12.0,6.5hz,h-6β),6.89(1h,s,h-2’),6.89(1h,s,h-6’),6.83(1h,s,h-2”),6.83(1h,s,h-6”),6.17(1h,d,j=2.3hz,h-3
”’
),6.10(1h,d,j=2.3hz,h-5
”’
),2.53(3h,s,h-8
”’
),3.80(3h,s,och3);
13
c nmr(125mhz,dmso-d6):δ
c 96.7(c-1),73.3(c-2),74.9(c-3),67.7(c-4),77.1(c-5),60.4(c-6),119.1(c-1’),108.8(c-2’),145.5(c-3’),138.6(c-4’),145.5(c-5’),108.8(c-6’),165.1(c-7’),118.5(c-1”),108.8(c-2”),145.5(c-3”),139.0(c-4”),145.5(c-5”),108.8(c-6”),164.7(c-7”),106.9(c-1
”’
),162.7(c-2
”’
),92.0(c-3”),162.7(c-4
”’
),96.2(c-5
”’
),165.2(c-6
”’
),203.2(c-7
”’
),32.9(c-8
”’
),56.2(och3)。
[0082][0083]
实施例5
[0084]
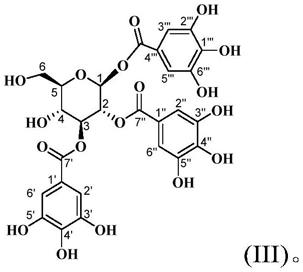
本发明化合物1,2,3-tri-o-galloyl-β-d-glucopyranose的其分子结构式如下:
[0085][0086]
1,2,3-tri-o-galloyl-β-d-glucopyranose的物理性质为淡白色无定型粉末;核磁数据如下:
[0087]
uv(meoh)λ
max 215,280nm;hr-esi-ms:m/z 635.0889[m-h]-(cal.635.0890);1h nmr(500mhz,dmso-d6):δ
h 6.05(1h,d,j=8.3hz,h-1),5.41(1h,dd,j=9.9,8.2hz,h-2),5.55(1h,t,j=9.5hz,h-3),3.73-3.97(4h,m,h-4,h-5,h-6),7.03(1h,s,h-2’),7.02(1h,s,h-6’),7.03(1h,s,h-2”),7.02(1h,s,h-6”);
13
c nmr(125mhz,dmso-d6):δ
c 93.9(c-1),72.4(c-2),76.8(c-3),69.3(c-4),79.1(c-5),61.9(c-6),119.9(c-1’),110.0(c-2’),146.4(c-3’),146.6(c-4’),146.4(c-5’),110.4(c-6’),166.4(c-7’),120.5(c-1”),110.0(c-2”),146.4(c-3”),146.4(c-4”),146.4(c-5”),110.4(c-6”),167.2(c-7”),121.1(c-1
”’
),110.4(c-2
”’
),146.6(c-3”),146.6(c-4
”’
),146.6(c-5
”’
),110.6(c-6
”’
),167.8(c-7
”’
)。
[0088]
实施例6
[0089]
本实施例提供了一种以euphorbiacetophenone e为原料药的片剂,其组份如下:
[0090][0091][0092]
制成100片。
[0093]
取euphorbiacetophenone e与羟丙基甲基纤维素、滑石粉、乳糖、硬脂酸镁混匀后,加入无水乙醇制成软材,过24目筛,制成颗粒,干燥,加入硬脂酸镁,混匀,压片。
[0094]
实施例7
[0095]
本实施例提供了一种以1,2,3-tri-o-galloyl-β-d-glucopyranose为原料药的片剂,其组份如下:
[0096]
1,2,3-tri-o-galloyl-β-d-glucopyranose24.0mg羟丙基甲基纤维素16g滑石粉0.3g乳糖0.2g硬脂酸镁0.2g无水乙醇0.1ml
[0097]
制成100片。
[0098]
取1,2,3-tri-o-galloyl-β-d-glucopyranose与羟丙基甲基纤维素、滑石粉、乳糖、硬脂酸镁混匀后,加入无水乙醇制成软材,过24目筛,制成颗粒,干燥,加入硬脂酸镁,混匀,压片。
[0099]
实施例8
[0100]
本实施例提供了一种以化合物euphorbiacetophenone e为原料药的胶囊剂,其组份如下:
[0101][0102][0103]
制成100粒。
[0104]
取euphorbiacetophenone e与淀粉、微晶纤维素、焦亚硫酸氢钠混匀后,加入无水乙醇制成软材,过24目筛,制成颗粒,干燥,加入硬脂酸镁,混匀,装入胶囊。
[0105]
实施例9
[0106]
本实施例提供一种以化合物1,2,3-tri-o-galloyl-β-d-glucopyranose为原料药的胶囊剂,其组份如下:
[0107]
1,2,3-tri-o-galloyl-β-d-glucopyranose19.0mg微晶纤维素0.2g淀粉5.0g焦亚硫酸氢钠0.2g硬脂酸镁0.2g
无水乙醇0.1ml
[0108]
制成100粒。
[0109]
取1,2,3-tri-o-galloyl-β-d-glucopyranose与淀粉、微晶纤维素、焦亚硫酸氢钠混匀后,加入无水乙醇制成软材,过24目筛,制成颗粒,干燥,加入硬脂酸镁,混匀,装入胶囊。
[0110]
实施例10
[0111]
本实施例提供一种以化合物euphorbiacetophenone e为原料药的颗粒剂,其组份如下:
[0112][0113][0114]
制成100袋。
[0115]
取euphorbiacetophenone e与淀粉、亚硫酸氢钠混匀后,加入无水乙醇制成软材,过24目筛,制成颗粒,干燥,加入硬脂酸镁,混匀,装袋。
[0116]
实施例11
[0117]
本实施例公开一种以化合物1,2,3-tri-o-galloyl-β-d-glucopyranose为原料药的颗粒剂,其组份如下:
[0118]
1,2,3-tri-o-galloyl-β-d-glucopyranose34.0mg淀粉5g焦亚硫酸氢钠0.2g硬脂酸镁0.2g无水乙醇0.1ml
[0119]
制成100袋。
[0120]
取1,2,3-tri-o-galloyl-β-d-glucopyranose与淀粉、亚硫酸氢钠混匀后,加入无水乙醇制成软材,过24目筛,制成颗粒,干燥,加入硬脂酸镁,混匀,装袋。
[0121]
实施例12
[0122]
本实施例公开一种以化合物euphorbiacetophenone e为原料药的口服液,其组份如下:
[0123][0124][0125]
制成100支。上述组分混匀后,采用口服液常规制备方法,分装即可。
[0126]
实施例13
[0127]
本实施例提供了一种以化合物1,2,3-tri-o-galloyl-β-d-glucopyranose为原料药的口服液,其组份如下:
[0128]
1,2,3-tri-o-galloyl-β-d-glucopyranose24.0mg蔗糖2.0g亚硫酸氢钠0.2g对羟基苯甲酸甲酯0.2g碳酸氢钠0.1ml注射用水1000ml
[0129]
制成100支。上述组分混匀后,采用口服液常规制备方法,分装即可。
[0130]
实施例14
[0131]
本实施例提供了一种以化合物euphorbiacetophenone e为原料药的注射剂,其组份如下:
[0132]
euphorbiacetophenone e45.0mg维生素c0.2g氯化钠6.0g碳酸氢钠0.1ml注射用水1000ml
[0133]
制成100支。上述组分混匀后,采用注射剂常规制备方法,即可得100支。
[0134]
实施例15
[0135]
本实施例公开一种以化合物1,2,3-tri-o-galloyl-β-d-glucopyranose为原料药的注射剂,其组份如下:
[0136]
1,2,3-tri-o-galloyl-β-d-glucopyranose44.0mg维生素c0.2g氯化钠5.0g碳酸氢钠0.1ml
注射用水1000ml
[0137]
制成100支。上述组分混匀后,采用注射剂常规制备方法,即可得100支。
[0138]
实施例16
[0139]
本实施例提供了一种以化合物euphorbiacetophenone e和阿卡波糖为原料药的片剂,其组份如下:
[0140]
euphorbiacetophenone e20.0mg阿卡波糖5g羟丙基甲基纤维素18g滑石粉0.4g乳糖0.2g硬脂酸镁0.2g无水乙醇0.1ml
[0141]
制成100片。取euphorbiacetophenone e、阿卡波糖和羟丙基甲基纤维素、滑石粉、乳糖、硬脂酸镁混合均匀,加无水乙醇制成软材,过24目筛,制成颗粒,干燥,加入硬脂酸镁,混匀,压片。
[0142]
实施例17
[0143]
本实施例公开一种以化合物1,2,3-tri-o-galloyl-β-d-glucopyranose和阿卡波糖为原料药的片剂,其组份如下:
[0144][0145][0146]
制成100片。
[0147]
取1,2,3-tri-o-galloyl-β-d-glucopyranose、阿卡波糖和羟丙基甲基纤维素、滑石粉、乳糖、硬脂酸镁混合均匀,加无水乙醇制成软材,过24目筛,制成颗粒,干燥,加入硬脂酸镁,混匀,压片。
[0148]
实施例18
[0149]
本实施例提供一种以化合物euphorbiacetophenone e和伏格列波糖为原料药的胶囊剂,其组份如下:
[0150]
euphorbiacetophenone e18.0mg
伏格列波糖2.0g微晶纤维素0.2g淀粉6.0g焦亚硫酸氢钠0.2g硬脂酸镁0.2g无水乙醇0.1ml
[0151]
制成100粒。
[0152]
取euphorbiacetophenone e、伏格列波糖和淀粉、微晶纤维素、焦亚硫酸氢钠混匀后,加入无水乙醇制成软材,过24目筛,制成颗粒,干燥,加入硬脂酸镁,混匀,装入胶囊。
[0153]
实施例19
[0154]
本实施例提供了一种以化合物1,2,3-tri-o-galloyl-β-d-glucopyranose和伏格列波糖为原料药的胶囊剂,其组份如下:
[0155]
1,2,3-tri-o-galloyl-β-d-glucopyranose17.0mg伏格列波糖2.0g微晶纤维素0.2g淀粉5.0g焦亚硫酸氢钠0.2g硬脂酸镁0.2g无水乙醇0.1ml
[0156]
制成100粒。
[0157]
取1,2,3-tri-o-galloyl-β-d-glucopyranose、伏格列波糖和淀粉、微晶纤维素、焦亚硫酸氢钠混匀后,加入无水乙醇制成软材,过24目筛,制成颗粒,干燥,加入硬脂酸镁,混匀,装入胶囊。
[0158]
实施例20
[0159]
本实施例公开一种以化合物euphorbiacetophenone e和胰岛素为原料药的注射剂,其组份如下:
[0160]
euphorbiacetophenone e40.0mg胰岛素2.0g维生素c0.2g氯化钠6.0g碳酸氢钠0.1ml注射用水1000ml
[0161]
制成100支。上述组分混匀后,采用注射剂常规制备方法,即可得100支。
[0162]
实施例21
[0163]
本实施例公开一种以化合物1,2,3-tri-o-galloyl-β-d-glucopyranose和胰岛素为原料药的注射剂,其组份如下:
[0164]
1,2,3-tri-o-galloyl-β-d-glucopyranose39.0mg胰岛素2.0g
维生素c0.2g氯化钠5.0g碳酸氢钠0.1ml注射用水1000ml
[0165]
制成100支。上述组分混匀后,采用注射剂常规制备方法,即可得100支。
[0166]
实施例6至实施例21所制备的药物制剂均对α-葡萄糖苷酶具有明显的抑制作用,可以作为治疗糖尿病疾病中的药物进行应用。
[0167]
为了更好地理解本发明的实质,下面结合药理试验及结果来说明上述糖苷类化合物在制药领域中的新应用。
[0168]
试验例1
[0169]
本试验例提供化合物euphorbiacetophenone e、1,2,3-tri-o-galloyl-β-d-glucopyranose及已知化合物3,5-dihydroxy-2,4-dimethyl-1-o-(6'-o-galloyl-1-β-d-glucopyranol)-benzophenone、salaciacochinoside a、3,4,6-tri-o-galloyl-β-d-glucose和1-o-gallicacy-6-o-(3-methoxy)caffeicacyl-β-d-glucopyranose的α-葡萄糖苷酶抑制活性试验。
[0170]
(1)实验材料和仪器
[0171]
α-葡萄糖苷酶(来源于酵母),4-硝基苯-α-d-吡喃葡萄糖苷(p-npg),阿卡波糖均购自上海源叶生物科技有限公司;pbs(北京索莱宝科技有限公司);试剂无水na2co3为分析纯。
[0172]
二氧化碳细胞培养箱(美国thermo fisher scientific公司);全波长酶标仪(美国thermo fisher scientific公司);移液器(德国eppendorf公司)。
[0173]
(2)实验方法
[0174]
实验分为空白组、空白对照组、样品空白组和样品组。实验在96孔板中进行,样品组及样品空白组中每孔加入不同浓度的样品或阳性药阿卡波糖20μl,每组设置3个平行复孔,分别加入100μl pbs缓冲溶液,空白对照组和样品空白组加入120μl pbs缓冲溶液,空白组加入140μl pbs缓冲溶液;样品组及空白对照组组加入20μlα-葡萄糖苷酶磷酸缓冲溶液(0.25u/ml),37℃孵育15min后取出,加入20μl 2.5mmol/lp-npg溶液,充分混匀,37℃孵育15min,结束后加入40μl 0.2mol/l的na2co3溶液中止反应。在405nm处测定吸光值,根据以下公式:
[0175]
抑制率(%)=[(od control-od control blank)-(od sample-od sample blank)]/(od control-od control blank)
×
100%
[0176]
计算出各样品对α-葡萄糖苷酶的抑制率。以阿卡波糖为阳性对照,求出相应的ic
50
值。
[0177]
(3)结果,实验结果如表1所示:
[0178]
表1.euphorbiacetophenone e、1,2,3-tri-o-galloyl-β-d-glucopyranose等化合物对α-葡萄糖苷酶抑制活性筛选
[0179][0180]
表1的结果表明糖苷类化合物euphorbiacetophenone e及1,2,3-tri-o-galloyl-β-d-glucopyranose对于α-葡萄糖苷酶具有良好的抑制作用,抑制活性均优于阳性药阿卡波糖及其他已知化合物,抑制作用由大到小排列为1,2,3-tri-o-galloyl-β-d-glucopyranose>euphorbiacetophenone e>3,4,6-tri-o-galloyl-β-d-glucose>3,5-dihydroxy-2,4-dimethyl-1-o-(6'-o-galloyl-1-β-d-glucopyranol)-benzophenone>acarbose>1-o-gallicacy-6-o-(3-methoxy)caffeicacyl-β-d-glucopyranose>salaciacochinoside a,表明本发明所述的糖苷类化合物是天然产物中α-葡萄糖苷酶抑制活性较强的分子,因此可以作为治疗糖尿病疾病中的药物应用。
[0181]
试验例2
[0182]
本试验例公开一种化合物1,2,3-tri-o-galloyl-β-d-glucopyranose对α-葡萄糖苷酶抑制动力学试验。
[0183]
(1)实验材料和仪器
[0184]
α-葡萄糖苷酶(来源于酵母),4-硝基苯-α-d-吡喃葡萄糖苷(p-npg),阿卡波糖均购自上海源叶生物科技有限公司;pbs(北京索莱宝科技有限公司);试剂无水na2co3为分析纯。
[0185]
二氧化碳细胞培养箱(美国thermo fisher scientific公司);全波长酶标仪(美国thermo fisher scientific公司);移液器(德国eppendorf公司)。
[0186]
(2)α-葡萄糖苷酶结合方式的确定
[0187]
按照“试验1”方法分别测定不同浓度1,2,3-tri-o-galloyl-β-d-glucopyranose(0,12.5,25,50μmol/l)与α-葡萄糖苷酶浓度分别为0.125、0.25、0.5、1.0u/ml时反应的初速度,底物p-npg最终浓度为2.5mmol/l,每个浓度做三个平行孔,以酶浓度e(u/ml)为横坐标,反应初速度ν(δod/min)为纵坐标作图,利用图的特征推断酶的结合方式。
[0188]
(3)α-葡萄糖苷酶抑制类型的确定
[0189]
按照“试验1”方法分别测定不同浓度1,2,3-tri-o-galloyl-β-d-glucopyranose(0,12.5,25,50μmol/l)与底物p-npg浓度分别为0.625、1.25、2.5、5mmol/l时反应的初速度,加入α-葡萄糖苷酶浓度为0.25u/ml,每个浓度做三个平行孔,按照michaelis-menten及lineweaver-burk方程进行统计作图,绘制抑制作用动力学曲线,确定抑制类型。
[0190]
(4)统计分析
[0191]
数据采用graphpad prism 6.02软件处理,所有实验结果均测量3次,以平均值
±
标准差(x
±
sd)表示。
[0192]
(5)结果
[0193]
如图1及图2表明化合物1,2,3-tri-o-galloyl-β-d-glucopyranose为抑制活性良好的α-葡萄糖苷酶抑制剂,且以与酶非共价键结合使其活力丧失或降低,为可逆抑制剂
[0194]
由图3及图4可知,化合物1,2,3-tri-o-galloyl-β-d-glucopyranose与α-葡萄糖苷酶的抑制类型为混合型抑制,随着底物浓度的增大,v
max
减小,km增大,即该化合物不仅能与底物竞争与酶活性中心结合,还能与游离酶或酶-底物复合物结合从而使酶活力降低或丧失。
[0195]
综上,本发明所述的糖苷类化合物具有优于降糖一线用药阿卡波糖的抑制活性,可以作为治疗糖尿病疾病中的药物应用。
[0196]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。