1.本发明属于化合物领域,具体涉及一类硅保护4-氨基的杂环类化合物2的直接金属化反应及其与不同亲电试剂r4lg反应,用于制备嘌呤核苷类似物式3的方法及应用。
背景技术:
2.嘌呤核苷类似物是重要的生物活性化合物,在抗肿瘤和抗病毒药物开发中具有重要意义。大量嘌呤核苷类似物显示了抗肿瘤、抗病毒、抗凝血等生物活性,其中瑞德西韦、阿昔洛韦、恩替卡韦、替格瑞洛等嘌呤核苷类似物已经上市。
3.嘌呤类似物式2的7位修饰较为困难,因此相应的嘌呤核苷类似物也就难以获得。通常需要将结构式2a的4-氨基杂环类化合物上的7位卤代后,交换为碱金属【organ ic chemi stry front iers 2018,5,1992-1999.】或碱土金属【1)
4.bioorganic&medicinal chemistry letters 2012,22(12),4127-4132.2)journal of medicinal chemistry 2014,57(5),1812-1825.】,最后再与亲电试剂反应得到式5结构的5a和5b,具体如下:
[0005][0006][0007][0008]
以上基于嘌呤类似物式2的嘌呤核苷类似物的合成方法均有收率低于60%或路线
较长等问题。严重影响了工业化大生产。
技术实现要素:
[0009]
本发明是从易于制备、成本较低的式2a的4-氨基杂环类化合物出发,经由4-nh2临时硅保护,避免使用7-卤素取代步骤,即可直接将7-位金属化,并合成式3化合物,具有收率高、区域选择性好和易于工业化生产等显著的优势。
[0010]
本发明的技术方案如下:
[0011]
一种杂环化合物,其具有以下式3化合物的结构:
[0012][0013]
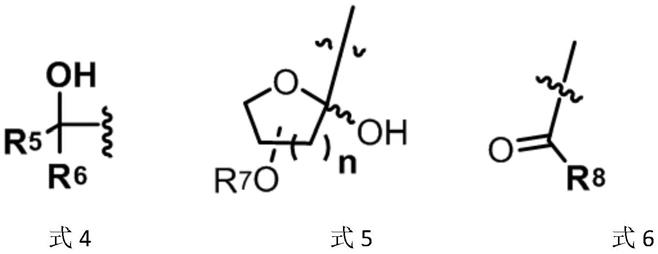
式3化合物中:r4选自含取代基的c1~c12的饱和或不饱和碳链,或选自如结构式4所示的羟基化合物结构,或选自如结构式5所示的n=1~2的含环内氧原子的环状结构,或选自如结构式6所示的r8(c=o)-片段;
[0014][0015]
其中r5、r6分别选自h、含取代基的c1~c12的饱和或不饱和的碳链、含取代基的芳香基,
[0016]
r7为硅烷保护基、苄基保护基、取代苄基保护基、甲氧基甲基保护基,
[0017]
r8选自含取代基的芳香基团或含取代基的c1~c12的饱和或不饱和碳链。
[0018]
优选方案为:所述式3化合物的结构式具体为如下式的式3a、3b、3c、4a、5a或5c:
[0019]
[0020]
本发明的另一个发明目的是提供上述杂环化合物的制备方法,其包括如下步骤:
[0021]
(1)以下面式2a的4-氨基杂环类化合物为原料,将式2a中的4-氨基在碱的存在下,用卤代硅烷保护得式2化合物,
[0022][0023]
其中r为单硅烷保护氨基、双硅烷保护氨基或环二硅烷保护氨基;
[0024]
(2)将亲电试剂r4’lg溶解,或将r4’lg与稀土盐混合,得亲电试剂液;
[0025]
(3)在有机金属试剂存在的条件下,在温度为-20~-100℃,选择性脱除式2化合物7位上h后,再与步骤(2)中的亲电试剂液反应得到式3化合物,
[0026][0027]
当式3化合物中r4选自结构式4所示的羟基化合物结构时,r4’lg为r5(c=o)r6结构;
[0028]
或当r4选自结构式5所示的n=1~2的含环内氧原子的环状结构时,r4’lg为内酯结构;
[0029]
或当r4选自其他结构时,r4’lg为环氧化物;或r4’lg中的r4’同r4、lg选自卤素、甲磺酰氧基、对甲苯磺酰氧基或三氟甲磺酰氧基。
[0030]
进一步方案,步骤(1)中所述的碱选自三乙胺、n,n,n',n'-四甲基乙二胺、n,n-二甲基苯胺、甲基锂、正丁基锂、叔丁基锂、仲丁基锂、苯基锂、甲基卤化镁、乙基卤化镁、苯基卤化镁、异丙基卤化镁、异丙基卤化镁配合物或氢化钠;
[0031]
所述卤代硅烷为三甲基氯硅烷、三乙基氯硅烷、叔丁基二甲基氯硅烷、亚乙基四甲基二硅烷二氯化物;
[0032]
先将式2a化合物与溶剂混合形成悬浮液,再加入卤代硅烷混合;然后滴加碱,得式2化合物。
[0033]
进一步方案,步骤(2)中稀土盐为三氯化镧、三氯化铈或三氯化钕;所述r4’lg与稀土盐的摩尔比为1:0.5-2。
[0034]
进一步方案,步骤(3)中是在温度为-40~-70℃下选择性脱除式2化合物7位上h;
[0035]
式2化合物和有机金属试剂的摩尔比1:1~3,反应得到式3化合物的温度为-40~-70℃;
[0036]
所述有机金属试剂为烷基或芳基锂试剂、烷基镁试剂、叔丁醇钠、或叔丁醇钾。
[0037]
进一步方案,所述烷基或芳基锂试剂为甲基锂、正丁基锂、叔丁基锂、仲丁基锂、苯基锂;所属烷基镁试剂包括异丙基氯化镁、异丙基氯化镁-氯化锂复合物、2-甲基丙基氯化
镁、2-甲基丙基氯化镁-氯化锂复合物、2,2,6,6-四甲基哌啶基氯化镁-氯化锂复合物。
[0038]
进一步方案,r4’lg作为内酯时,其结构包括:
[0039][0040]
当r4’lg作为内酯时,对应加成产物为如下式的式5a、式5b、式5c:
[0041][0042]
本发明的第三个发明目的是提供上述的一种杂环化合物的应用,所述式3化合物作为制备c-核苷衍生物的中间体。
[0043]
进一步方案,所述c-核苷衍生物的中间体为如下式的式5aa、式5cc、式7、式8、或式5ab化合物:
[0044][0045]
所以,本发明的合成路线如下:
[0046][0047]
式2中r为单硅烷保护氨基、双硅烷保护氨基或环二硅烷保护氨基,结构式如下:
[0048][0049]
式3化合物为新颖结构的嘌呤核苷类似物,可用于生物活性研究。其中含有式4、式5或式6结构的式3化合物可用于合成多种新型c-核苷衍生物的中间体,该中间体如下式:
[0050][0051]
式3化合物的结构还可以合成如下式的式5b结构,式5b还可经历已知的中间体7和8而合成瑞德西韦9,因而提供了一种更有效的瑞德西韦合成方法:
[0052][0053]
本发明具有以下有益效果:
[0054]
以化合物5b合成为例,现有制备工艺中是以如式2a的4-氨基杂环类化合物的卤代反应,易于生成5,7-二卤代衍生物,所以收率通常不高,如碘代反应,其收率为70-80%,经碘代中间体的糖苷化反应收率在40-60%之间【journal of medicinal chemistry 2014,
57(5),1812-1825,doi:10.1021/acs.oprd.0c00172】,所以两步合并收率一般在35-45%之间。由于式2a化合物的价格昂贵,低收率导致生产成本显著提高。而本技术是将式2a化合物直接金属化,从而减少了7-卤素取代这一步骤,简化了制备方法,并且本技术中基于式2a化合物的直接金属化和c-核苷加成的收率达到近80%有显著的成本优势。
[0055]
其次,为了提高金属交换效率,通常需要采用碘来取代式2a化合物成7-碘代式2a化合物,而碘每千克的价格高达200元,所以昂贵的碘取代又增加了成本。即便使用7-碘代式2a化合物进行金属交换反应,其与亲电试剂r4’lg反应的收率也较低。由于对于大多数r4’lg结构,除了羰基反应中心与式2a的金属化衍生物反应以外,羰基中心的alpha-h也会竞争性地与式2a的金属化衍生物反应,从而降低加成收率。
[0056]
本技术选用适当的碱来促进式2a中的4-nh2进行卤代硅烷保护,然后在温度为-20~-100℃下,用有机金属试剂(如正丁基锂)直接脱除式2化合物7-h。最后,对亲电试剂r4’lg进行加成反应;当r4’lg存在羰基alpha位活泼氢时,稀土盐的存在可以促进此加成反应的收率。
[0057]
即本技术采用成本较低的式2a的4-氨基杂环类化合物出发,经由4-nh2临时硅保护,避免使用7-卤素取代步骤,即可直接将式2a化合物7-位金属化,而合成式3化合物,具有收率高、区域选择性好和易于工业化生产等显著的优势。其总收率显著高于文献的7-卤代方案。
具体实施方式
[0058]
下面结合实施例对本发明作进一步说明。
[0059]
实施例1
[0060][0061]
(1)将式2a化合物(1.0mmol)悬浮于溶剂thf(四氢呋喃)(5ml)中,室温条件下加入tmscl(三甲基氯硅烷)(1.1mmol),随后降温至-60℃,滴加过量的正丁基锂(2.5m己烷溶液,3.2mmol),滴毕后保持在-60℃下搅拌2h;制得式2b化合物的同时脱除式2b化合物7位上h;
[0062]
(2)另外准备一个反应瓶,加入亲电试剂环氧乙烷(2.0mmol)和thf(10ml),室温搅拌均匀得亲电试剂液;
[0063]
(3)将步骤(2)制得的亲电试剂液预冷至-60℃后加入步骤(1)中,继续在-60℃搅拌3h;然后逐步热至-20℃,加入醋酸(3mmol)淬灭反应,
[0064]
(4)将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物式3a化合物标准样品,收率79%,硅胶薄层色谱比移值0.35(二氯甲烷:甲醇=10:1),h-nmr(400mhz,dmso-d6)δ7.77(s,1h),7.57(brs,2h),6.78(d,1h),6.43(d,1h),4.71(m,1h),3.65(m,2h),2.98(t,2h);esi-ms:201.3(m na离子)。
[0065]
实施例2
60℃,并将上述锂化混合物加入此反应液中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物式3a化合物,收率69%,分析数据与化合物3a标准样品相同。
[0079]
实施例5
[0080][0081]
将式2a化合物(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(2.4mmol),随后降温至-60℃,滴加过量的正丁基锂(2.5m己烷溶液,3.8mmol),滴毕后-60℃搅拌2h;制得式2c化合物的同时脱除式2c化合物7位上h;
[0082]
另外准备一个反应瓶,加入环氧乙烷(2.0mmol)和thf(10ml),并将此溶液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物式3a化合物,收率62%,分析数据与化合物3a标准样品相同。
[0083]
实施例6
[0084][0085]
将式2a化合物(1.0mmol)悬浮于thf(5ml)中,室温下加入1,2-双(氯二甲基硅基)乙烷(1.1mmol),随后降温至0℃,加入nah(60%悬浮于矿物油中,2.1mmol),随后在0℃搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,1.1mmol),滴毕后-60℃搅拌2h;制得式2d化合物的同时脱除式2d化合物7位上h;。
[0086]
另外准备一个反应瓶,加入环氧乙烷(2.0mmol)和thf(10ml),并将此溶液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物3a,收率69%,分析数据与化合物3a标准样品相同。
[0087]
实施例7
[0088][0089]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温
至0℃,加入nah(60%悬浮于矿物油中,1.1mmol),随后在0℃搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0090]
另外准备一个反应瓶,加入苄氯(2.0mmol)和thf(10ml),并将此溶液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物3b标准样品,收率90%,硅胶薄层色谱比移值0.8(二氯甲烷:甲醇=10:1),h-nmr(400mhz,dmso-d6)δ7.80(s,1h),7.48-7.59(m,1h),7.32-7.11(m,5h),6.80(d,1h),6.41(d,1h),4.05(s,2h);esi-ms:247.5(m na离子)。
[0091]
实施例8
[0092][0093]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至0℃,加入nah(60%悬浮于矿物油中,1.1mmol),随后在0℃搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0094]
另外准备一个反应瓶,加入溴乙酸乙酯(2.5mmol)和thf(10ml),将此溶液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物3c,收率82%,硅胶薄层色谱比移值0.5(二氯甲烷:甲醇=10:1),h-nmr(400mhz,dmso-d6)δ8.28(bs,1h),8.10(m,2h),7.25(d,j=4.7hz,1h),7.00(d,j=4.6hz,1h),4.15(q,j=6.9hz,3h),3.78(s,2h),1.27(t,j=6.9hz,4h).esi-ms:243.4(m na离子)。
[0095]
实施例9
[0096][0097]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至0℃,加入n,n-二异丙基乙胺(2.0mmol),随后在0度搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0098]
另外准备一个反应瓶,加入s-(-)-甘油醛缩丙酮(2.0mmol)、thf(10ml)、三氯化钕(2.0mmol)和无水四丁基氯化铵(2.0mmol),室温搅拌5h,将此悬浊液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物4a标准样品,收率49%,硅胶薄层色谱比移值0.5(二氯甲烷:甲醇=10:1),esi-ms:287.3(m na离子)。
[0099]
实施例10
[0100][0101]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至0℃,加入n,n-二异丙基乙胺(2.0mmol),随后在0度搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0102]
另外准备一个反应瓶,加入s-(-)-甘油醛缩丙酮(2.0mmol)、thf(10ml)、三氯化镧(2.2mmol)和无水四丁基氯化铵(2.0mmol),室温搅拌5h,将此悬浊液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物4a,收率42%,分析数据与实施例8中的化合物4a标准样品相同。
[0103]
实施例11
[0104][0105]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至0℃,加入二异丙基乙基胺(2.0mmol),随后在0度搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0106]
另外准备一个反应瓶,加入2,3,4,6-四-o-苄基-d-葡萄糖-δ-内酯(1.2mmol)、thf(10ml)、三氯化钕(1.2mmol)和无水四丁基氯化铵(1.2mmol),室温搅拌5h,将此反应液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5a,收率67%,硅胶薄层色谱比移值0.6(二氯甲烷:甲醇=10:1),esi-ms:655.9(m-oh正离子)。
[0107]
对比例1
[0108][0109]
将化合物2a(1.0mmol)溶于dmf(5ml)中,冷却至0℃,分批加入n-碘代丁二酰亚胺(1.0mmol),0℃继续搅拌3h,随后在40℃以下浓缩反应液,再加入水析出固体,过滤并水洗得到产物2a-i,收率75%,esi-ms:283.3(m na正离子),核磁数据与文献【cn108348526a】一致。
[0110][0111]
将化合物2a-i(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至0℃,加入n,n-二异丙基乙胺(2.0mmol),随后在0度搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0112]
另外准备一个反应瓶,加入2,3,4,6-四-o-苄基-d-葡萄糖-δ-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol)和无水四丁基氯化铵(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5a,收率20%,分析数据与化合物5a标准样品相同。
[0113]
对比例1与实施例11对比发现:本技术实施例10制备的化合物5a的收率为67%,而对比例1制备的化合物5a的收率为20%,所以本技术的制备方法不仅减少了7-卤素取代这一步骤,简化了制备方法,而且还显著提高了最终产品的收率。
[0114]
实施例12
[0115][0116]
将实施例10制备的化合物5a(1mmol)溶于dcm(5ml)中,冷却至0℃,加入三乙基硅烷(2.5mmol),继续搅拌10min,再加入三氟化硼乙醚(1.2mmol)。将此混合物逐步热至室温,继续搅拌1h,加入饱和碳酸氢钠水溶液并分液,将有机相用饱和食盐水洗涤、浓缩并柱层析得到产品5aa,收率82%,esi-ms:679.9(m na正离子)。
[0117]
实施例13
[0118][0119]
将上述实施例10制备的化合物5a(1mmol)在dcm(5ml)中的溶液冷却至-35℃,加入三氟乙酸(3mmol),再加入tmsotf(6mmol)和tmscn(6mmol)在dcm(5ml)中预冷至-40℃的混合溶液,随后将此混合物在-30℃搅拌30min,并加入预冷至-10℃的koh水溶液中终止,将此混合物分液后,有机相用饱和食盐水洗涤、浓缩、并柱层析得到产物5ab,收率43%,esi-ms:
705.0(m na正离子)。
[0120]
实施例14
[0121][0122]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至0℃,加入n,n-二异丙基乙胺(2.0mmol),随后在0度搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0123]
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol)和无水四丁基氯化铵(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5b标准样品,收率79%,硅胶薄层色谱比移值0.6(二氯甲烷:甲醇=10:1),esi-ms:535.8(m-oh正离子),核磁数据与文献一致【doi:10.1038/nature17180】。
[0124]
实施例15
[0125][0126]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至0℃,加入n,n-二异丙基乙胺(2.0mmol),随后在0度搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0127]
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol)和无水四丁基氯化铵(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,并将上述锂化混合物加入此反应液中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5b标准样品,收率77%,分析数据与5b标准样品一致。
[0128]
对比例2
[0129]
[0130]
将对比例1制备的化合物2a-i(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至0℃,加入n,n-二异丙基乙胺(2.0mmol),随后在0℃搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0131]
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol)和无水四丁基氯化铵(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5b,收率27%,分析数据与化合物5b标准样品相同。
[0132]
对比例2与实施例14对比发现:本技术实施例13制备的化合物5b的收率为79%,而对比例2制备的化合物5b的收率为27%。所以本技术的制备方法不仅减少了7-卤素取代这一步骤,简化了制备方法,而且还显著提高了最终产品的收率。
[0133]
实施例16
[0134][0135]
将实施例13制备的化合物5b(1mmol)在dcm(5ml)中的溶液冷却至-40℃,加入三氟乙酸(3mmol),再加入tmsotf(6mmol)和tmscn(6mmol)在dcm(5ml)中预冷至-40℃的混合溶液,随后将此混合物在-30℃搅拌30min,并加入预冷至-10℃的koh水溶液中终止,将此混合物分液后,有机相用饱和食盐水洗涤、浓缩、并柱层析得到产物7,收率72%,分析数据与文献【doi:10.1021/acs.oprd.0c00172】一致。
[0136][0137]
将上述制备的化合物7(1mmol)溶于dcm(5ml)中冷却至-20℃,加入三氯化硼(1m dcm溶液,3.4mmol),继续在-20℃搅拌3h,随后加入甲醇(30ml),将此混合物在-10度以下浓缩至无溶剂流出,再加甲醇(30ml)浓缩至无溶剂流出,随后将此混合物加入碳酸钾水溶液(20%),逐步热至室温,析出固体,过滤,水洗,并干燥,得到产物8,收率80%,分析数据与文献【doi:10.1038/nature17180】一致。
[0138]
实施例17
[0139]
[0140]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至0℃,加入n,n-二异丙基乙胺(2.0mmol),随后在0度搅拌30min,再降温至-60℃,滴加2,2,6,6-四甲基哌啶基氯化镁-氯化锂(1.0m四氢呋喃-甲苯溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0141]
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol)和无水四丁基氯化铵(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5b,收率53%,分析数据与化合物5b标准样品相同。
[0142]
实施例18
[0143][0144]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至0℃,加入nah(60%悬浮于矿物油中,1.1mmol),随后在0度搅拌30min,再降温至-60℃,滴加正丁基锂(2.5m己烷溶液,2.2mmol),滴毕后-60℃搅拌2h。
[0145]
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol)和无水四丁基氯化铵(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5b,收率52%,分析数据与化合物5b标准样品相同。
[0146]
实施例19
[0147][0148]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至-60℃,滴加正丁基锂(2.5m己烷溶液,3.2mmol),滴毕后-60℃搅拌2h。
[0149]
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol)和无水四丁基氯化铵(1.1mmol),室温搅拌过夜,将此反应液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5b,收率75%,分析数据与化合物5b标准样品相同。
[0150]
实施例20
[0151][0152]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至-60℃,滴加正丁基锂(2.5m己烷溶液,3.2mmol),滴毕后-60℃搅拌2h。
[0153]
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化镧(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5b,收率60%,分析数据与化合物5b标准样品相同。
[0154]
实施例21
[0155][0156]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至-60℃,滴加正丁基锂(2.5m己烷溶液,3.2mmol),滴毕后-60℃搅拌2h。
[0157]
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-核糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化铈(1.1mmol),室温搅拌24h,将此反应液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5b,收率43%,分析数据与化合物5b标准样品相同。
[0158]
实施例22
[0159][0160]
将化合物2a(1.0mmol)悬浮于thf(5ml)中,室温下加入tmscl(1.1mmol),随后降温至-60℃,滴加正丁基锂(2.5m己烷溶液,3.2mmol),滴毕后-60℃搅拌2h。
[0161]
另外准备一个反应瓶,加入2,3,5-三苄氧基-d-阿拉伯糖酸-1,4-内酯(1.1mmol)、thf(10ml)、三氯化钕(1.1mmol),室温搅拌5h,将此反应液预冷至-60℃,加入上述锂化混合物中。将此混合物继续在-60℃搅拌3h,逐步热至-20℃,加入醋酸(3mmol)淬灭反应,再将反应液依次用水、饱和碳酸氢钠水溶液、以及饱和食盐水洗涤。最后将所得有机相浓缩后柱层析得到产物5c,收率75%,硅胶薄层色谱比移值0.47(二氯甲烷:甲醇=10:1),h-nmr
(400mhz,dmso-d6)8.05(s,1h),7.86(s,1h),7.81(s,1h),7.64(s,1h),7.3~7.1(m,10h),6.95(m,1h),6.71(m,1h),5.34(m,1h),4.65(m,6h),4.71(m,1h),2.5(m,2h);esi-ms:429.4(m-oh正离子)。
[0162]
实施例23
[0163][0164]
将上述制备的化合物5c(1mmol)溶于dcm(5ml)中,冷却至0℃,加入三乙基硅烷(2.5mmol),继续搅拌10min,再加入三氟化硼乙醚(1.2mmol)。将此混合物逐步热至室温,继续搅拌1h,加入饱和碳酸氢钠水溶液并分液,将有机相用饱和食盐水洗涤、浓缩并柱层析得到产品5cc,收率70%,esi-ms:453.4(m na正离子)。
[0165]
本发明显示并详细描述的信息足以实现本发明的上述目的,因此,本发明的范围不被除所附权利要求之外的任何内容所限制,其中除了明确说明外,所用元素的单数形式并不是指“一个和唯一”,而是指“一个或更多”。对本领域一般技术人员来说,所有公知的上述优选的实施方案和附加实施方案部分的结构、组成和功能上的等价物因此引入本文作参考,而且试图被本发明的权利要求所涵盖。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。