trilaciclib的合成方法
技术领域
1.本技术属于合成化学技术领域,尤其涉及一种trilaciclib的合成方法。
背景技术:
2.2021年2月,食品药品监督管理局(fda)批准了新型cdk4/6抑制剂cosela(trilaciclib)的上市申请,该药物隶属于g1 therapeutics公司旗下,是一款降低广泛期小细胞癌症患者在接受某些类型化疗时出现的骨髓抑制的药物,也是首款获批用于该适应症的cdk4/6抑制剂。cosela(trilaciclib)可通过抑制靶标cdk4/6的活性来保护骨髓细胞免受化疗引起的损伤,做为一种短效cdk4/6抑制剂,患者在接受化疗前使用该药,可让骨髓干细胞发生短期的细胞周期停滞,从而让骨髓干细胞免受化疗药物的伤害。
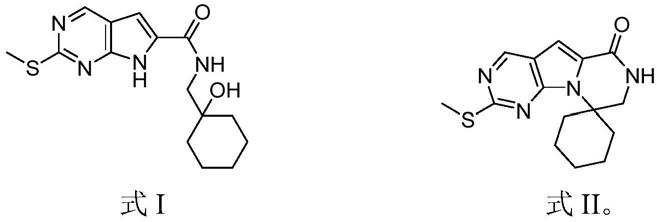
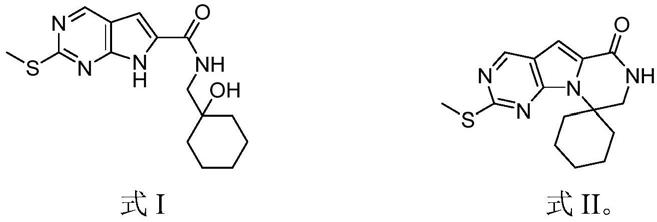
3.trilaciclib的结构式如下,其游离碱的cas号为:1374743
‑
00
‑
6;
[0004][0005]
目前,该trilaciclib的合成可采用原研公司g1 therapeutics公司开发的方法:(一)以4
‑
氯
‑2‑
甲硫基嘧啶
‑5‑
羧酸乙酯为起始原料,经亲核取代、boc保护酰胺基、分子内环化、酚羟基上ts、脱除ots、氧化硫醚、脱除保护基boc得到甲砜嘧啶并吡咯衍生物,甲砜嘧啶并吡咯衍生物在强碱作用下,与1
‑
甲基
‑4‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪发生亲核取代反应,得到最终产品trilaciclib;(二)以2,4
‑
二氯
‑5‑
溴嘧啶为起始原料,经亲核取代、sonogashira反应、分子内环化、缩醛脱保护、醛氧化为酸、脱除保护基boc、分子内酰胺化得到氯代嘧啶并吡咯衍生物,氯代嘧啶并吡咯衍生物同样在强碱作用下,与1
‑
甲基
‑4‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪发生亲核取代反应,得到最终产品trilaciclib。上述两种合成方法工艺路线较长,总收率较低。
技术实现要素:
[0006]
本技术的目的在于提供一种trilaciclib的合成方法,旨在解决trilaciclib合成路线长、收率低的技术问题。
[0007]
为实现上述申请目的,本技术采用的技术方案如下:
[0008]
本技术提供一种trilaciclib的合成方法,包括如下步骤:
[0009]
将2
‑
(甲硫基)
‑
7h
‑
吡咯[2,3
‑
d]嘧啶
‑6‑
羧酸和1
‑
氨基甲基
‑1‑
环己醇进行缩合反应,得到式i所示的中间体1:
[0010]
将所述中间体1进行内环化反应,得到式ii所示的中间体2;
[0011]
将所述中间体2与1
‑
甲基
‑4‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪进行亲核取代反应,得到trilaciclib;
[0012][0013]
本技术提供的trilaciclib合成方法,以2
‑
(甲硫基)
‑
7h
‑
吡咯[2,3
‑
d]嘧啶
‑6‑
羧酸和1
‑
氨基甲基
‑1‑
环己醇为原料,进行缩合反应得到中间体1,然后将中间体1进行分子内环化得到中间体2,最后将中间体2与1
‑
甲基
‑4‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪进行反应,得到trilaciclib;本技术的合成方法相对现有方法显著缩短了合成路线,提高了总收率,因此,在trilaciclib制备领域具有很好的应用前景。
具体实施方式
[0014]
为了使本技术要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本技术进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本技术,并不用于限定本技术。
[0015]
本技术中,术语“和/或”,描述关联对象的关联关系,表示可以存在三种关系,例如,a和/或b,可以表示:单独存在a,同时存在a和b,单独存在b的情况。其中a,b可以是单数或者复数。字符“/”一般表示前后关联对象是一种“或”的关系。
[0016]
本技术中,“至少一种”是指一种或者多种,“多种”是指两种或两种以上。“以下至少一项(个)”或其类似表达,是指的这些项中的任意组合,包括单项(个)或复数项(个)的任意组合。
[0017]
应理解,在本技术的各种实施例中,上述各过程的序号的大小并不意味着执行顺序的先后,部分或全部步骤可以并行执行或先后执行,各过程的执行顺序应以其功能和内在逻辑确定,而不应对本技术实施例的实施过程构成任何限定。
[0018]
在本技术实施例中使用的术语是仅仅出于描述特定实施例的目的,而非旨在限制本技术。在本技术实施例和所附权利要求书中所使用的单数形式的“一种”、“所述”和“该”也旨在包括多数形式,除非上下文清楚地表示其他含义。
[0019]
本技术实施例说明书中所提到的相关成分的重量不仅仅可以指代各组分的具体含量,也可以表示各组分间重量的比例关系,因此,只要是按照本技术实施例说明书相关组分的含量按比例放大或缩小均在本技术实施例说明书公开的范围之内。具体地,本技术实施例说明书中所述的质量可以是μg、mg、g、kg等化工领域公知的质量单位。
[0020]
术语“第一”、“第二”仅用于描述目的,用来将目的如物质彼此区分开,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。例如,在不脱离本技术实施例范围的情况下,第一xx也可以被称为第二xx,类似地,第二xx也可以被称为第一xx。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。
[0021]
本技术实施例所使用缩写的含义列于下表中:
[0022]
表1
[0023][0024]
[0025]
本技术实施例提供一种trilaciclib的合成方法,该合成方法包括如下步骤:
[0026]
s01:以2
‑
(甲硫基)
‑
7h
‑
吡咯[2,3
‑
d]嘧啶
‑6‑
羧酸和1
‑
氨基甲基
‑1‑
环己醇为原料,进行缩合反应,得到式i所示的中间体1:
[0027]
s02:将中间体1进行内环化反应,得到式ii所示的中间体2;
[0028]
s03:将中间体2与1
‑
甲基
‑4‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪进行亲核取代反应,得到trilaciclib;
[0029][0030]
本技术实施例提供的trilaciclib合成方法,具体路线如下,以2
‑
(甲硫基)
‑
7h
‑
吡咯[2,3
‑
d]嘧啶
‑6‑
羧酸和1
‑
氨基甲基
‑1‑
环己醇为原料,进行合反应得到中间体1,然后将中间体1进行分子内环化得到中间体2,最后将中间体2与1
‑
甲基
‑4‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪进行反应,得到trilaciclib;本技术实施例的合成方法相对现有方法显著缩短了合成路线,提高了总收率,因此,在trilaciclib制备领域具有很好的应用前景。
[0031][0032]
步骤s01中:为2
‑
(甲硫基)
‑
7h
‑
吡咯[2,3
‑
d]嘧啶
‑6‑
羧酸和1
‑
氨基甲基
‑1‑
环己醇的缩合反应,具体地,可以在缩合剂的条件下进行缩合反应,通过缩合剂的作用使两者缩合得到式ii所示的中间体1。其中,缩合剂选自hobt、edc.hcl、hatu、dcc、dic、dipea、hbtu、hoat、hobt、pybop、pyaop、tbtu、btc中的一种或多种;进一步地,优选的缩合剂为hobt和edc.hcl的组合。
[0033]
该缩合反应可以在第一溶剂中进行,具体地,第一溶剂选自n,n
‑
二甲基甲酰胺、四氢呋喃、2
‑
甲基四氢呋喃、丙酮、1,4
‑
二氧六环、乙腈、甲苯和二氯甲烷中的一种或多种。缩合反应的温度为
‑
80℃~200℃,具体地,可以为0~200℃,或50~200℃,进一步地,缩合反应温度可以为100~150℃,反应时间可以为10~25h。
[0034]
在一个实施例中,上述缩合反应的具体步骤包括:将2
‑
(甲硫基)
‑
7h
‑
吡咯[2,3
‑
d]嘧啶
‑6‑
羧酸和1
‑
氨基甲基
‑1‑
环己醇溶于第一溶剂中,然后加入上述缩合剂,在上述温度
条件下进行缩合反应。
[0035]
步骤s02中:为中间体1进行分子内环化反应,具体地,该内环化反应在催化剂条件下进行,经催化剂催化分子内环化得到式ii所示的中间体2。其中,催化剂选自pph3、pme3、addp、dead、diad、cmmp、cmbp、phsih3、phi(oac)2中的一种或多种。
[0036]
该内环化反应可以在第二溶剂中进行,具体地,第二溶剂选自n,n
‑
二甲基甲酰胺、六氟异丙醇、三氟乙酸、四氢呋喃、2
‑
甲基四氢呋喃、丙酮、1,4
‑
二氧六环、乙腈、甲苯和二氯甲烷中的一种或多种。内环化反应的温度为
‑
30℃~220℃,具体地,可以为0~220℃,或50~220℃,进一步地,内环化反应温度可以为100~160℃,反应时间可以为20~35h。
[0037]
在一个实施例中,上述内环化反应的具体步骤包括:将上述中间体1溶于第二溶剂中,然后加入上述催化剂,在上述温度条件下进行催化内环化反应。
[0038]
步骤s03中:为trilaciclib的生成步骤,将中间体2与1
‑
甲基
‑4‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪进行亲核取代反应,得到目标产物trilaciclib。具体地,该亲核取代反应在高温无溶剂条件下进行。
[0039]
亲核取代反应的温度为80℃~300℃,具体可以为100~300℃,或200~300℃。进一步地,亲核取代反应的温度为200~250℃,反应时间为1~4h。
[0040]
在一个实施例中,上述亲核取代反应的具体步骤包括:将上述中间体2与1
‑
甲基
‑4‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪混合于真空管中,然后在上述温度条件下进行反应。反应结束后,用水和饱和食盐水依次洗涤,然后干燥过滤,去溶剂得到目标产物trilaciclib。
[0041]
下面结合具体实施例进行说明。
[0042]
实施例1
[0043]
式i所示的中间体1的制备
[0044][0045]
于500ml三颈瓶中依次加入dmf 150ml、dcm 150ml、2
‑
(甲硫基)
‑
7h
‑
吡咯[2,3
‑
d]嘧啶
‑6‑
羧酸21g(100mmol)、1
‑
氨基甲基
‑1‑
环己醇14.3g(110mmol)、hobt 15.2g(100mmol)、edc.hcl 19.2g(100mmol),100℃反应20小时。反应结束后,体系中加入1n稀碳酸氢钠,调ph值=7~8,用dcm萃取,合并有机相,用饱和食盐水洗涤有机相,用无水硫酸镁干燥,将所得滤液减压浓缩,得到棕色粗产物。在所得粗产物中加入300ml异丙醇重结晶,得到25.7g类白色固体产物,即得到中间体1,纯度为99%,收率为85%。
[0046]
所得到的产物经质谱和核磁共振确认结构,结果为:1h nmr(400mhz,dmso
‑
d6)δ1.30
‑
1.65(m,10h),2.48(s,3h),3.10(d,2h),7.11(s,1h),8.81(s,1h),11.90(br,1h).lcms(esi):[m h
]321.42.
[0047]
实施例2
[0048]
式i所示的中间体1的制备
[0049]
于500ml三颈瓶中依次加入dmf 150ml、dcm 150ml、2
‑
(甲硫基)
‑
7h
‑
吡咯[2,3
‑
d]
嘧啶
‑6‑
羧酸21g(100mmol)、1
‑
氨基甲基
‑1‑
环己醇14.3g(110mmol)、tbtu 32.1g(100mmol)、dipea 12.9g(100mmol),120℃反应20小时。反应结束后,体系中加入1n稀碳酸氢钠,调ph值=7~8,用dcm萃取,合并有机相,用饱和食盐水洗涤有机相,用无水硫酸镁干燥,将所得滤液减压浓缩,得到棕色粗产物。在所得粗产物中加入320ml异丙醇重结晶,得到25.4g类白色固体产物,即得到中间体1,纯度为99%,收率为84%。
[0050]
实施例3
[0051]
式ii所示的中间体2的制备
[0052][0053]
中间体1(25.7g,80mmol)和四氢呋喃
‑
甲苯混合溶剂(体积比为1:2,总量为120ml)加至500ml三颈瓶中,0℃滴加cmbp(38.6g,160mmol),搅拌30min,然后升温至150℃,反应35小时。反应结束后,加入饱和柠檬酸溶液,调ph值为7.5,分出体系的有机相,水相用甲苯(80ml
×
3次)萃取,合并有机相,用饱和盐水(120ml)洗涤,用无水硫酸钠干燥后过滤,将所得滤液减压蒸除溶剂,得到黄色粘稠状粗品22g。将此粗品溶于热异丙醚中,然后滴加到冰正己烷中,出现白色固体后立即过滤,用正己烷洗涤数次,干燥,得到白色纯品中间体2(21.1g),纯度为95%,收率为87%。
[0054]
所得到的产物经质谱和核磁共振确认结构,结果为:1h nmr(400mhz,dmso
‑
d6)δ1.31
‑
1.70(m,10h),2.47(s,3h),3.15(d,2h),7.10(s,1h),8.90(s,1h).lcms(esi):[m h
]303.40.
[0055]
实施例4
[0056]
式ii所示的中间体2的制备
[0057]
中间体1(25.7g,80mmol)和四氢呋喃
‑
甲苯混合溶剂(体积比为1:2,总量为120ml)加至500ml三颈瓶中,0℃滴加cmmp(18.4g,160mmol),phsih3(17.3g,160mmol),搅拌30min,然后升温至160℃,反应35小时。反应结束后,加入饱和柠檬酸溶液,调ph值为7.5,分出体系的有机相,水相用甲苯(80ml
×
3次)萃取,合并有机相,用饱和盐水(120ml)洗涤,用无水硫酸钠干燥后过滤,将所得滤液减压蒸除溶剂,得到黄色粘稠状粗品22g。将此粗品溶于热异丙醚中,然后滴加到冰正己烷中,出现白色固体后立即过滤,用正己烷洗涤数次,干燥,得到白色纯品中间体2(20.6g),纯度为95%,收率为85%。
[0058]
实施例5
[0059]
trilaciclib的制备
[0060][0061]
将中间体2(21.1g,70mmol)和1
‑
甲基
‑4‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪(13.4g,70mmol)
加入到100ml真空管中230℃反应3h。反应结束后,加入乙酸乙酯300ml,依次用水(120ml)和饱和食盐水(120ml)洗涤,用无水硫酸钠干燥后过滤,将所得溶液减压蒸除溶剂,所得剩余物用乙醇重结晶,得到trilaciclib27.5g,纯度为99%,收率为88%。
[0062]
所得到的产物经质谱和核磁共振确认结构,结果为:1h nmr(400mhz,dmso
‑
d6)δ1.27
‑
1.44(m,10h),1.79
‑
1.87(m,5h),2.62
‑
2.69(m,2h),3.16
‑
3.36(m,4h),3.63
‑
3.73(m,2h),7.11(s,1h),7.30(d,1h),7.69(d,1h),7.87(dd,1h),8.81(s,1h).lcms(esi)(m h)447.
[0063]
以上所述仅为本技术的较佳实施例而已,并不用以限制本技术,凡在本技术的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本技术的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。