一种超微量全长rna测序文库的构建方法
技术领域
1.本发明属于生物技术领域,涉及一种超微量全长总rna测序文库构建方法,尤其涉及一种基于模板转换逆转录及crispr/cas9高效切割去除rrna,同时能够通过编码进行细胞或者亚细胞样本并行测序与分析的超微量的全长总rna转录组建库方法。
背景技术:
2.最近,随着高通量测序技术的发展,单细胞rna测序技术(single cell rna sequencing,scrna
‑
seq)成为可能。2009年,tang等人发表了第一个单细胞rna
‑
seq测序方案。但由于测序通量低,随后strt
‑
seq和scrb
‑
seq作为新的方法被引入,它们可以同时处理多个不同的样本,但通常会引入3
′
端或5
′
端的偏差,与之相比,smart
‑
seq2结合模板转换方法,进行了全转录组的测序,可用于融合基因检测、单核苷酸变异(snv)分析、可变剪切等,成为了一种单细胞全长转录组测序分析的理想方法。此外,为了减少上述方法中pcr扩增所产生的偏差,cel
‑
seq和mars
‑
seq方法使用体外逆转录(ivt)替代pcr扩增,获得足够多的cdna量进行测序,且降低了pcr扩增偏差。最近,基于液滴和分裂池连接的方法能够获得数千个单细胞,为解析细胞异质性和稀有细胞类型提供的新的可能。但所有的这些方法的不足都是通过oligo
‑
dt的方法进行mrna及少量长链非编码rna的富集,而其它的非编码rna很难获取。这就限制了我们对非编码rna的深入解析,成为解析单个细胞中全部转录信息的主要障碍。
3.目前,研究者们在努力开发单细胞全转录组rna
‑
seq方法,如最早的super
‑
seq,利用特异性随机引物富集非polya的rna,包括circrnas。然而,super
‑
seq对非polya尾巴的rna敏感性相对较低(20%
‑
30%)。这为研究scrna
‑
seq测序非poly(a)尾rnas的富集方法提供了空间。此外,一个不可忽视的问题就是在总rna测序中,不感兴趣的rna物种丰度(如rrna占细胞总量的80%
‑
90%)会占据测序的容量,影响其它低丰度转录物的结果分析,同时还增加了测序的成本。目前,从总rna中去除rrna的方法包括两种,直接富集多聚腺苷酸化(polya)的转录本和靶向去除rrna。前者主要由于rrna无polya尾,因此可以使用oligo(dt)引物富集含polya尾的mrna,也由于操作步骤简单方便而成为了大多数scrna
‑
seq富集mrna的主要方法,包括smart
‑
seq2/3、cel
‑
seq2等。然而,这种方法很容易产生偏差,因为它去除了除rrna之外所有的非编码转录本,如长的非编码rna(lncrna)、3
′
末端降解的mrna等。另一种方法,rrna特异性去除方法,通过使用生物素标记的特异性探针(如illumina’s ribo
‑
zero和thermo fisher’s ribominus)或rnase h介导的降解(如:neb’s nebnext)。虽然这些靶向去除的方法保留了大部分的非rrna,但往往需要较高的样本投入量10ng
‑
1μg,远高于单细胞rna量的要求,很难在scrna
‑
seq中应用,从而限制了研究者对单细胞全转录组信息的分析。
4.因此为了以最高效率从scrna
‑
seq文库中去除rrna,研究者们提出了在cdna合成过程中或者合成之后进行rrna的去除,从而降低了对rna输入量的要求。目前最具代表性的方法为以takara为代表的sczapr and scr
‑
probes,可在单细胞建库中高效率的去除rrna,
但其价格及其昂贵。此外随着crispr技术的日益成熟,研究者开发了一种新的通过利用crispr/cas9技术杂交去除非靶标序列(dash),其原理是cas9核酸酶与single guide rnas(sgrnas)形成复合物,在特定sgrna互补位置诱导双链断裂(dsbs),从而去除靶基因,如rrna。此外,研究者也应用crispr/cas9从atac
‑
seq文库中切割去除线粒体dna。
5.目前,现有技术中无法准确的对样本转录组中不带poly(a)尾的rna进行测序的问题,尤其是无法去除细胞或者亚细胞样本转录组中的rrna,亟待出现一种方法能够解决上述问题。
技术实现要素:
6.发明目的:本发明所要解决的技术问题是提供了一种能够进行超微量全长总rna建库,同时能够高效去除rrna。
7.技术方案:为了实现上述目的,本技术采用了以下技术方案:一种超微量全长总rna测序文库的构建方法,所述构建方法主要包括以下步骤:
8.1)对获得的细胞或亚细胞样本中的超微量总rna,按照常规的方法进行cdna文库的构建,获得包含rrna序列信息的cdna文库;
9.2)对步骤1)中获得的cdna文库进行扩增;
10.3)根据细胞或亚细胞的相应物种的rrna序列,设计特异性的sgrna序列组合,所述的sgrna序列组合包括seq id no.1
‑
seq id no.58;
11.4)将sgrna序列组合配置的溶液与步骤2)扩增的cdna文库进行混合,利用crispr/cas9系统在cas9蛋白作用下进行特异性的切割,获得不包含rrna信息的cdna文库。
12.其中,所述步骤1)细胞或者亚细胞的rna的起始量为0.5~500pg。
13.其中,所述步骤2)的文库扩增可采用pcr或等温扩增。
14.其中,所述步骤4)中配置rrna的sgrna混合池及cas9的rnp复合物的反应体系,37℃孵育0.5
‑
2h。
15.其中,所述步骤4)中cas9蛋白的浓度为10nm
‑
2μm。
16.其中,所述步骤4)中sgrna序列组合的浓度为0.1
‑
1μm。
17.其中,所述超微量全长转录组测序文库的构建方法具体包括如下步骤:
18.s1)将细胞或者亚细胞裂解获得rna;
19.s2)rna片段化、逆转录及模板转换:将rna置于二价阳离子溶液、带有修饰和编码的半随机引物、dntp、第一链合成试剂的混合液中进行片段化,其后在模板转换引物、rna酶抑制剂、smartscirbe逆转录酶、dtt、甜菜碱及片段化的产物进行模板置换合成二链cdna;
20.s3)将得到的二链cdna预扩增及pcr产物纯化;
21.s4)二轮pcr扩增及二轮pcr扩增产物纯化及片段筛选;
22.s5)利用crispr/cas9系统进行特异性的切割,获得不包含rrna信息的cdna文库;
23.s6)cdna文库上机测序。
24.其中,所述步骤s3)预扩增反应循环数是15~24,所述步骤4)第二轮反应循环数为18~25。
25.其中,所述步骤s2)的带有修饰和编码的半随机引物序列为:biotin
‑5′‑
gtctcgtgggctcggagatgtgtataagagacagxxxxxxxxnnnnnsn
‑3′
;其中x代表的是编码序列。
26.其中,所述步骤s2)的模板转换引物序列为:biotin
‑5′‑
tcgtcggcagcgtcagatgtgtataagagacagrgrgrg
‑3′
。
27.其中,所述步骤s3)的预扩增的扩增引物包括ispcr
‑
oligo引物和ispcr
‑
tso引物,所述ispcr
‑
oligo引物序列为:gtctcgtgggctcggagatgtgtataagagacag;所述ispcr
‑
tso引物序列为:tcgtcggcagcgtcagatgtgtataagagacag。
28.其中,所述步骤s4)的二轮pcr扩增使用p5引物(引物序列:aatgatacggcgaccaccgagatctacactcgtcggcagcgtc)、p7引物(引物序列:caagcagaagacggcatacgagatyyyyyyyygtctcgtgggctcgg;y为index序列,长度为6~8bp)和2x kapa的高保真酶进行pcr扩增反应。
29.其中,所述步骤s5)根据相应物种的rrna序列,设计了如下sgrna的引物序列:
30.表1 sgrna序列
31.[0032][0033]
本发明的一种能够进行超微量全长总rna建库方法,在rna到cdna的逆转录过程中所使用的引物包含随机引物和特定序列,随机引物位于特定序列的3’端,二者之间有1~5个碱基,随机序列由3~18个碱基组成,优选6~15个随机碱基组成(a、t、g、c任意合成六到十五聚体),特定序列为8~45个碱基的同聚体,优选15~32个碱基的同聚体,特定序列的碱基选自a、t、g、c中的任意一个。常用的oligo
‑
dt作为引物的方法中,由于引物只能结合在mrna链末端的聚腺苷酸尾巴上,导致绝大部分情况下能够覆盖的片段都在整条核糖核酸链的末尾段,不利于完整转录组的研究及揭示可变剪切等现象。本发明的半随机引物理论上可以结合整条核糖核酸链的各个部位,因此可以大大提高逆转录的均匀性,反应细胞中转录本的真实状态。然后利用pcr等扩增方法进行微量cdna的放大;针对待去除的rrna基因的cds区域设计得到sgrna(single guide rna)pool,使cdna文库样本与sgrna pool、cas9蛋白接触,使其对相应的cdna序列进行切割,最后获得除核糖体rna以外的所有细胞rna序列全转录组测序文库。在该方法中,所述建库过程较常规转录组建库的优势在于可以进行低至~0.5pg的样本建库,同时能够实现rna的全长测序和rrna的高效去除。
[0034]
在该方法中,逆转录之前使用化学方法(如mg
2
,zn
2
等)在70~90℃将rna打断,反应时间2~25min,优选5~20min,更优选10min。rna通常具有丰富的二级结构,在常规的方法中,逆转录前对rna在65℃条件下加热5min使rna的二级结构充分打开,实际上较为稳定
的二级结构在这种条件下并不能完全打开。因此本方法通过优化精确控制加热温度和时间,使rna的二级结构充分打开,从而增加了逆转录效率。在该方法中,提供了一整套rrna的sgrna序列,弥补了其它技术方案仅有线粒体或者细胞质的rrna的sgrna序列。在该方法中,提供了一整套完整的编码信息,克服超微量rna全长转录组单管测序的障碍,可实现大量样本的并行测序与分析。
[0035]
本发明提供了一整套完整的超微量全长总rna建库流程。得到的样品直接用于核酸文库构建操作,所以需要达到一定的文库构建起始量,需要进行两轮的pcr反应,第一轮预扩增反应循环数是15~24,第二轮反应循环数为18~25,需要根据实验起始量的大小(总的核苷酸含量多少)决定。以10pg核糖核酸起始量为例,第一轮和第二轮pcr扩增分别需要20和25个循环。本方法适用于样本起始量低至~0.5pg的实验,虽然测序结果显示,随着样本投入量的增加,基因的覆盖度显著增加,但ng级别的样本起始量已经达到饱和,过度的扩增会带来更大的偏差,超过500ng的样本起始量得到的实验结果的稳定性可能反而不如100ng。
[0036]
使用本发明的超微量全长总rna建库方法拥有很高的可重复性,适于分析单细胞及微量样本中全长转录组信息,从而可以更加全面的研究单细胞或微量样本的全长转录组,对更多未知的核糖核酸展开研究。同时,本发明可以克服超微量全长转录组单管测序的障碍,实现大量样本的并行测序与分析。
[0037]
本发明的该方法使用包含随机序列的“半随机引物”进行逆转录,不依赖polya尾,因此可以覆盖所有核糖核酸种类;同时在逆转录之后使用crispr/cas9进行高效的rrna的去除,解决了超低量样本和单细胞样本投入量的需求。
[0038]
本发明采用了带有编码和修饰的半随机引物进行逆转录,在cdna合成之后使用crispr/cas9技术从总rna中去除rrna,从而实现全长转录组建库,且同时能够通过编码进行大量样本的并行测序与分析,具有成本低廉,且能表征各种类型细胞转录本信息的差异性,非编码rna信息,为生物医学相关领域提供更加全面和精确的转录本信息。
[0039]
有益效果:与现有技术相比,本发明具有以下显著优势:
[0040]
(1)既能对带polya尾的rna进行测序,又能对不带polya尾的rna进行测序;
[0041]
(2)在cdna合成后使用crispr/cas9进行高效rrna的去除,rrna的比对率可低至1.5%,显著优于现有的试剂盒。
[0042]
(3)适用于超低起始量的转录组测序建库,rna的起始量可低至0.5pg。
[0043]
(4)可以克服多样本全长转录组单管测序的障碍,实现大量样本并行测序与分析。
[0044]
(5)可以实现rna的全长建库测序。
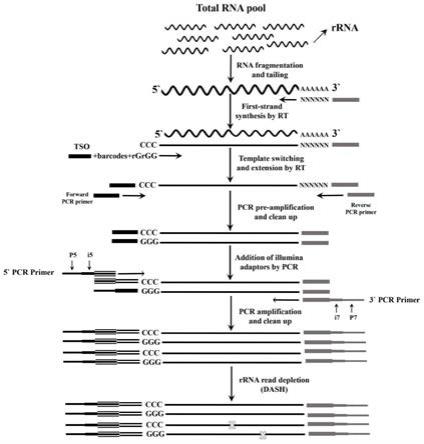
附图说明
[0045]
图1建库流程图;
[0046]
图2预扩增结果,样本1和样本2分别表示的从小鼠大脑区域获取的2个单细胞样本;
[0047]
图3pcr扩增结果,样本1和样本2分别表示的从小鼠大脑区域获取的2个单细胞样本。
[0048]
图4数据过滤的统计图,样本1和样本2分别表示的从小鼠大脑区域获取的2个单细
胞样本。adapter polluted reads rate(%):去掉含有接头污染的reads数占原始未过滤reads数的比例;ns reads rate(%):由于含n过高,被去掉的序列占原始下机序列的比例;low
‑
quality reads rate(%):被低质量过滤标准去掉的reads的比例;clean reads rate(%):过滤后剩余的reads数占原始未过滤reads数的比例,这个值越大,说明测序质量或者文库质量越好。
具体实施方式
[0049]
以下描述用于揭露本发明以使本领域技术人员能够实现本发明。以下描述中的实施例只作为举例,本领域技术人员可以想到其它显而易见的变型。在以下描述中界定的本发明的基本原理可以应用于其他实施方案、变形方案、改进方案、等同方案以及没有背离本发明的精神和范围的其他技术方案。
[0050]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
[0051]
下述实施例中所用材料、试剂等,如无特殊说明,均可从商业途径得到。
[0052]
本文使用的所有技术和科学术语具有与本领域技术人员通常理解的相同的含义。例如,术语“rna”是指单条rna链和多条rna链。
[0053]
本发明提供了超微量全长总rna测序文库构建方法。相对于现有的转录组文库构建方法,本发明可有效的去除rrna,获取样本全转录组信息。本发明超微量全长转录组rrna去除文库构建方法还对微量和降解样品中rna进行转录组文库构建和测序。进一步地,本发明超微量全长总rna测序文库构建方法的半随机引物反转录步骤使用了随机引物序列,提高了rna的捕获能力。此外本发明提供了一整套完整的编码信息,克服多样本全长转录组单管测序的障碍,实现大量样本并行测序与分析。同时本发明提供了一整套rrna的sgrna序列,弥补了其它技术方案仅有线粒体或者细胞质的rrna的sgrna序列。
[0054]
本发明的所有序列均由生工生物工程(上海)股份有限公司,sangon biotech(shanghai)合成。
[0055]
在一个具体的实施方案中,所有的操作在一个无酶的环境中进行。操作前对超净台进行彻底清洗,用rna酶去除试剂进行擦拭,实验前利用紫外灯照射半小时以上。
[0056]
实施例1
[0057]
本实例样本选择了小鼠脑的单细胞样本,使用从上海南方模式生物科技股份有限公司购买的1只健康小鼠,收集小鼠脑的样本对其进行石蜡切片,然后使用东南大学生科院的激光显微切割仪(北京安麦格贸易有限公司,lmd6),从小鼠脑样本的不同区域中分离单个细胞(命名为样本1和样本2),其后对样本1和样本2进行超微量全长总rna建库,。
[0058]
本实施例全长转录组文库构建包括小鼠脑单细胞样本制备、细胞裂解、rna片段化、逆转录(rt)反应、cdna扩增、crispr/cas9去除核糖体rna等一系列步骤。试剂加样操作和pcr操作按照以下说明进行。
[0059]
1、小鼠脑单细胞样本制备
[0060]
将制备好的石蜡切片样本置于pixcell显微激光切割系统10
×
10倍目前直视观察,重点观察细胞形态。找到细胞密集、形态较好、染色满意区域,装载细胞收集器;在10
×
20倍目镜下调节监视器,按照如下条件:power 100mv、duration 15.5s、spots size 15.5μm、current 8.0ma,每张切片平均8000shorts条件进行捕获,其后选择其中捕获的两个细胞
命名为样本1和样本2进行下述的文库构建。
[0061]
2、细胞裂解液的制备
[0062]
参照单细胞裂解试剂盒(invitrogen,#4458355)说明书,配置如下裂解液(表2),混合均匀,室温放置5min;加入0.25μl的单细胞终止液,室温放置2min,置于冰上。
[0063]
表2细胞rna裂解
[0064][0065]
3、rna片段化、逆转录及模板转换
[0066]
片段化和逆转录是指将rna(10pg)置于25mm的氯化镁,10μm的随机引物(引物序列:biotin
‑5′‑
gtctcgtgggctcggagatgtgtataagagacagxxxxxxxx nnnnnsn
‑3′
;引物合成:生工生物工程(上海)股份有限公司,sangon biotech(shanghai)co.,ltd.;n为随机碱基:a、t、c、g;x代表的是标签序列(barcode)),10mm的dntp(takara,#639132),5x smartscirbe一链合成buffer(takara,#639536)的混合液中,在80℃条件下反应5min,进行rna的片段化;其后在10μm的模板转换引物(tso)(biotin
‑5′‑
tcgtcggcagcgtcagatgtgtataagagacagrgrgrg
‑3′
;引物合成公司同上),40u/μl rnase酶抑制剂(takara,#2313a),100u/μl smartscirbe逆转录酶(takara,#639536),100μm的dtt(invitrogen,#18064
‑
014),5m的甜菜碱(sigma,#61962)及片段化的rna在42℃,90min;10循环(50℃,2min;42℃,2min);85℃,5min;4℃条件下进行一链合成和模板转化。
[0067]
表3逆转录反应体系
[0068][0069]
表4模板转换体系
[0070]
[0071][0072]
4、预扩增
[0073]
上述cdna合成之后,在其混合液中加入5μm引物ispcr
‑
oligo(引物序列:gtctcgtgggctcggagatgtgtataagagacag;引物合成公司同上)、5μm引物ispcr
‑
tso(引物序列:tcgtcggcagcgtcagatgtgtataagagacag;引物合成公司同上)和2x kapa的高保真酶(roach,#kk2631),在95℃,3min;18循环(98℃,20s;67℃,15s;72℃,3min);72℃,5min的条件下进行pcr扩增,增加cdna的浓度。此处尽可能少的使用pcr循环数,降低过多循环数带来的pcr扩增偏差)反应体系如表5所示:
[0074]
表5预扩增反应体系
[0075][0076]
5、上述pcr产物纯化
[0077]
1)向上述每个pcr产物中加入无酶水至总体积50μl,然后使用agencourt ampure xp磁珠(beckman coulter,beverly,usa,#a63880),进行纯化;
[0078]
2)涡旋振荡混匀agencourt ampure xp磁珠并吸取35μl体积至50μl pcr产物中,使用移液器轻轻吹打10次充分混匀。室温孵育5min;
[0079]
3)将反应管放置磁力架上5min,分离磁珠和液体;
[0080]
4)去除上清液,用200μl 80%的乙醇清洗2次,每次清洗30s。弃乙醇,使磁珠干燥。
[0081]
5)加入12.5μl的无酶水溶解,用移液器轻轻吹打10次充分混匀,室温孵育5min。
[0082]
6)放置磁力架上澄清,吸取10μl至新的pcr管。其中8μl用于下面的pcr扩增,另外2μl用于琼脂糖凝胶电泳(琼脂糖凝胶电泳结果如图2,样本1和样本2分别表示的从小鼠大脑区域获取的2个单细胞样本)。
[0083]
6、接头添加
[0084]
在上述纯化后的产物样本1和样本2中分别加入5μm的p5端通用引物(引物序列:aatgatacggcgaccaccgagatctacactcgtcggcagcgtc;合成公司同上)、5μm的p7端测序引物(样本1添加的引物序列:caagcagaagacggcatacgagattaaggcgagtctcgtgggctcgg;样本2添加的引物序列:caagcagaagacggcatacgagatcgtactaggtctcgtgggctcgg;合成公司同上)及2x kapa的高保真酶(roach,#kk2631),在98℃,45s;20循环(98℃,15s;60℃,30s;72℃,10s);72℃,1min的条件下进行pcr扩增。反应体系如表6所示:
[0085]
表6 pcr扩增反应体系
[0086][0087]
7、上述pcr产物纯化
[0088]
1)向上述每个pcr产物中加入无酶水至总体积50μl,然后使用agencourt ampure xp磁珠(beckman coulter,beverly,usa,#a63880),进行纯化;
[0089]
2)涡旋振荡混匀agencourt ampure xp磁珠并吸取37.5μl体积至50μl pcr产物中,使用移液器轻轻吹打10次充分混匀,室温孵育5min;
[0090]
3)将反应管放置磁力架上5min,分离磁珠和液体;
[0091]
4)去除上清液,用200μl 80%的乙醇清洗2次,每次清洗30s。弃乙醇,使磁珠干燥。
[0092]
5)加入15μl的无酶水溶解,用移液器轻轻吹打10次充分混匀,室温孵育5min。
[0093]
6)放置磁力架上澄清,吸取14μl至新的pcr管。其中12μl用于rrna的去除,另外2μl用于琼脂糖凝胶电泳(琼脂糖凝胶电泳结果如图3,样本1和样本2分别表示的从小鼠大脑区域获取的2个单细胞样本)。
[0094]
8、使用crispr/cas9技术进行rrna的去除
[0095]
crispr/cas9系统是通过rna介导的dna内切酶进行基因组编辑操作,它们分别是crispr rna(crrna)和反式作用crispr rna(trans
‑
acting crispr rna,tracrrna)。这两种rna可以被"改装"成一个向导rna(single
‑
guide rna,sgrna)。sgrna包括一段20bp左右的dna识别区与一段固定序列,其dna识别区与靶位点处碱基互补,引导cas9蛋白在结合区随机切割dna双链。
[0096]
1)设计rrna去除的crispr/cas9敲除文库
[0097]
从ncbi数据库下载45s rrna基因(登入号:18s:nr_003286.2;5.8s:nr_003285.2;28s:nr_003287.2)的cds序列,选择多个转录本共同的cds序列。利用sgrnacas9 3.0.5软件,结合在线设计网站http://crispr.mit.edu/及使用benchling在目标序列的正链和负链上设计sgrna,计算每个sgrna的特异性评分(脱靶评分)和效率评分(目标评分)。从18s、
5.8s和28s rrna基因7,096bp序列中筛选出出58个sgrna,长度为20bp,gc含量范围为40%~60%。完整的sgrna序列列表见表1(生工生物工程(上海)股份有限公司,sangon biotech(shanghai)合成)。
[0098]
2)sgrna的体外合成
[0099]
sgrna的体外合成通常有两种策略:一种为构建含特异性序列的转录质粒,另一种则使用合成的oligo退火延伸生成含t7的转录启动子的双链dna分子,进而再使用t7rna聚合酶进行体外转录,从而获得sgrna。利用合成的oligo直接制备sgrna,具有操作简单快速、可实现高通量等优势,因此体外合成sgrna成为本实验首选方案。应用haigene一步法sgrna合成试剂盒(海基生物科技有限公司,#d0601)获得的sgrna,为37℃过夜孵育的sgrna,以下设计的试剂均来自试剂盒提供,操作步骤如下:
[0100]
a:target sense oligo的设计:选取target dna序列pam(ngg)的5
′
端20bp进行sense oligo引物设计,sense oligo引物结构包含保护碱基(aagc)、t7promoter(ttctaatacgactcactatagg)、target dna片段(20bp)和互补片段(gttttagagctaga)。
[0101]
sense oligo设计如下:
[0102]
target sense oligo:
[0103]5’‑
aagcttctaatacgactcactatagg(n)
20
gttttagagctaga
‑3’
[0104]
转录得到的anti sgrna oligo靶序列如下:
[0105]
ctcagtatgatgcttctgagctgaaagcgtccatgaaggggctggggactgatgaggactctctcattgagattctgctcaaggaccaaccaggagctgcaggaaatcaacagagtctacaaggaaatgcaaccttcatttccctgctggagaaggacatgcaaccttcatttccctgctggtcgtttccgacacctggccacctggagacagtgattttgggcctattgaaaacacctg横线部分为设计的sgrna靶区域,根据sgrna靶区序列需要合成的target sense oligo入列为:
[0106]
target sense oligo:
[0107]5’‑
aagcttctaatacgactcactataggaccttcatttccctgcgttttagagctaga
‑3’
[0108]
anti sgrna oligo转录后获得的sgrna序列如下,其中:gg转录起始位点;粗体中的序列为grna区;下划线为crrna区。的序列为grna区;下划线为crrna区。
[0109]
b:转录sgrna体系的配制,反应体系如表8。
[0110]
表7转录sgrna体系的配制
[0111]
[0112][0113]
37℃反应过夜,转录sgrna产量为159ng/μl(孵育时间越长,sgrna产量越高);转录完成后向上述反应液中加入2μl的无酶水,其后37℃孵育15min去除dna模板。反应完毕后将其置于
‑
80℃冰箱中备用。
[0114]
c:cas9的rnp复合物的形成(crispr/cas9 rnp)
[0115]
通常来说crispr/cas9 rnp(核糖核蛋白复合物)是由sgrna及cas9蛋白组成。步骤b中合成的sgrna与cas9蛋白混合后,可去除靶基因序列(rrna)。之所以采用这种方法的原因,是脱靶效应会比较低,且不会出现完成dna整合的风险,更适合本实验。详细的反应体系如表9;
[0116]
表8 cas9的rnp复合物的形成体系
[0117][0118]
在37℃,20min。其后进行核糖体cdna的去除,操作步骤如下:
[0119]
d:rrna的去除
[0120]
①
添加第7)步纯化的cdna 1ng样本到上述10μl核糖核蛋白复合物的反应溶液中;
[0121]
②
另添加2x cas9 buffer,用无酶水将体系补足到20μl;37℃,90min;
[0122]
e:cas9蛋白去除及样本纯化
[0123]
①
向步骤d的溶液中添加1μl蛋白酶k(20mg/ml),37℃,15min;
[0124]
②
向上述产物中加入无酶水至总体积50μl,然后使用agencourt ampure xp磁珠(beckman coulter,beverly,usa,#a63880),进行纯化;
[0125]
③
涡旋振荡混匀agencourt ampure xp磁珠并吸取37.5μl体积至50μl pcr产物中,使用移液器轻轻吹打10次充分混匀,室温孵育5min;
[0126]
④
将反应管放置磁力架上5min,分离磁珠和液体;
[0127]
⑤
去除上清液,用200μl 80%的乙醇清洗2次,每次清洗30s。弃乙醇,使磁珠干燥;
[0128]
⑥
加入12μl的无酶水溶解,用移液器轻轻吹打10次充分混匀,室温孵育5min。
[0129]
⑦
放置磁力架上澄清,吸取10μl至新的pcr管,使用illumina hiseq x
‑
10(illumina inc.,san diego,ca,usa)的双末端2
×
150bp测序平台进行测序;
[0130]
9、测序数据分析
[0131]
1)原始数据的过滤
[0132]
由于测序得到的原始序列中,会含有测序接头序列及低质量序列。因此在序列比对之前,我们首先对原始数据进行过滤,得到高质量的clean reads(过滤后剩余的reads数),后续分析都基于clean reads。其中原始数据的处理通常包含以下三部分:去除接头污染的reads,去除低质量reads(reads中质量值q<19的碱基占总碱基的50%以上,对于上端
测序,若一端为低质量reads,则会去掉两端reads)等,其结果如图4所示。
[0133]
2)测序数据比对
[0134]
获得clean reads数据后,首先进行rrna的比对和剩余部分rrna的去除,从rnacentral上下载小鼠的rrna参考序列,使用hista2构建索引,其后进行rrna的比对及rrna序列的去除,其中rrna比对结果参见表10。
[0135]
使用去除rrna序列的数据进行mrna比对,流程如下:首先在hisat2官网下载小鼠参考基因组(https://genome
‑
idx.s3.amazonaws.com/hisat/mm10_genome.tar.gz tar
‑
zxvf mm10_genome.tar.gz),在gencode下载注释基因(http://ftp.ebi.ac.uk/pub/databases/gencode/gencode_mouse/release_m27/gencode.vm27.annotation.gtf.gz)进行比对和计数,其结果参见表10。
[0136]
最后检测鉴定非编码rna—环状rna(circrna)数量:首先使用bwa(比对方法记载如下文献中:li h,durbin r.fast and accurate short read alignment with burrows
–
wheeler transform.bioinformatics.2009;25(14):1754
‑
1760)进行比对,其后使用ciri2(比对方法记载如下文献中:gao y,wang j,zhao f.ciri:an efficient and unbiased algorithm for de novo circular rna identification.genome biology.2015;16(1):4)对circrna进行鉴定和计数,其结果参见表10。
[0137]
表10建库测序结果
[0138][0139][0140]
以上结果表明,本发明的超微量全长转录组建库方法可以在微量样本输入的条件下,有效去除rrna,同时能检测到较多的基因和非编码rna(如环状rna)数量。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。