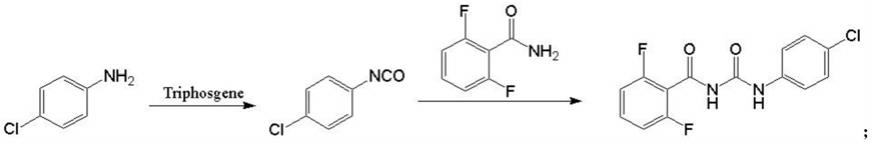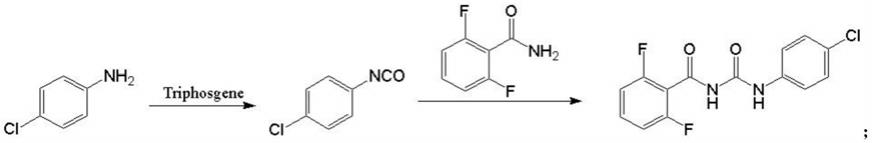
1.本发明属于农药的技术领域,具体涉及一锅法合成除虫脲的方法。
背景技术:
2.除虫脲属灭幼脲类杀虫剂,是20世纪70年代发现的昆虫生长调节剂,它的主要作用是抑制昆虫表皮的几丁质合成,同时对脂肪体、咽侧体等内分泌和腺体又有损伤破坏作用,使昆虫不能正常蜕皮虫体畸形而死亡,属低毒无公害农药。主要用于防治鳞翅目害虫,如菜青虫、小菜蛾、甜菜夜蛾、斜纹夜蛾、金纹细蛾、桃线潜叶蛾、柑橘潜叶蛾、粘虫、茶尺蠖、棉铃虫、美国白蛾、松毛虫、卷叶蛾、卷叶螟等。可广泛使用于苹果、梨、桃、柑橘等果树,玉米、小麦、水稻、棉花、花生等粮棉油作物,十字花科蔬菜、茄果类蔬菜、瓜类等蔬菜,及茶树、森林等多种植物。
3.目前所报道的除虫脲的合成方法有:1、以2,6
‑
二氟苯甲酰胺为起始原料,经酯化反应再与对氯苯胺缩合合成除虫脲;2、以对氯苯胺为起始原料,经酯化反应再与2,6
‑
二氟苯甲酰胺合成除虫脲。方法1中2,6
‑
二氟苯甲酰胺的酯化一般采用草酰氯作为酯化试剂(光气、三光气反应效果差,几乎不反应),价格昂贵,且储存和运输条件苛刻,工业化生产难度大。方法2中对氯苯胺酯化可选用固体光气作为酯化试剂,虽然三光气不论从成本上还是存储上都有优势,但目前所报道的合成方案中对氯苯胺酯化反应均会产生白色不溶杂质,需经过滤除去,酯化反应收率低,过滤设备成本高且过滤过程由于异氰酸酯本身味道大和反应残留的三光气会对环境造成很大的影响。同时对氯苯基异氰酸酯极怕水,遇水分解,熔点低(26
‑
29℃),在与溶剂分离过程会出现部分分解的情况,影响收率和品质从而影响除虫脲的合成成本,因此寻求一种催化剂促进异氰酸酯的合成的同时抑制杂质的生成,若能同时实现中间体异氰酸酯不出料一锅法直接进行下一步反应,将会有着较高的成本优势和工业化前景。
技术实现要素:
4.针对现有技术中合成除虫脲的方法存在的上述问题,本发明提供一锅法合成除虫脲的方法,以解决上述问题。
5.本发明的技术方案如下:
6.一锅法合成除虫脲的方法,合成路线如下:
[0007][0008]
具体步骤如下:
[0009]
(1)取三光气置于反应器中,加入溶剂ⅰ,搅拌溶清,降温;另取对氯苯胺,加入溶剂ⅱ溶清;控温滴加至三光气溶液中;
[0010]
(2)滴加完毕后,加入催化剂;然后开始梯度升温,保温反应;hplc监控反应;
[0011]
(3)反应结束后,向反应液中加入2,6
‑
二氟苯甲酰胺,然后升温至回流反应;取样hplc检测对氯苯异氰酸酯残留≤1%,反应结束;
[0012]
(4)降温析晶,得除虫脲粗品,粗品水洗,干燥,得除虫脲。
[0013]
优选的,所述对氯苯胺与三光气的投料摩尔比为1:0.34~0.50。
[0014]
优选的,所述溶剂ⅰ和溶剂ⅱ选自甲苯、二氯乙烷、二甲苯、氯苯、石油醚。
[0015]
优选的,所述催化剂为吡啶、3
‑
甲基吡啶、三乙胺、n,n
‑
二甲基甲酰胺、三苯基膦中的一种。
[0016]
优选的,所述催化剂用量为以对氯苯胺投料量计1~5wt%。
[0017]
优选的,所述2,6
‑
二氟苯甲酰胺与对氯苯胺的投料摩尔比为1~1.05:1。
[0018]
优选的,所述步骤(1)中,溶剂ⅰ的用量为以三光气投料量计2~4g/g;溶剂ⅱ的用量为以对氯苯胺投料量计6~12g/g。
[0019]
优选的,所述步骤(1)中,滴加对氯苯胺的温度为
‑
5~5℃。
[0020]
本发明的有益效果为:
[0021]
本发明所采用的催化剂有效的促进对氯苯胺异氰酸酯的合成,抑制杂质的产生。生成的异氰酸酯中间体无需分离提纯,可直接进行下步的合成,节省了分离提纯过程的同时,还降低了异氰酸酯中间体降解的风险。本发明具有工艺简单、副反应少、产品品质高、收率高、操作安全环保等优点,为工业化生产提供良好条件。
具体实施方式
[0022]
为了使本技术领域的人员更好地理解本发明中的技术方案,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
[0023]
实施例1
[0024]
(1)取12.11g(0.041mol)三光气置于500ml三口瓶中,加入36.33g二甲苯,搅拌溶清,降温至
‑
5~5℃;另取13.00g(0.102mol)对氯苯胺,加入91g二甲苯溶清,滴加至三光气溶液中;滴加过程控制反应体系温度为
‑
5~5℃;
[0025]
(2)滴加完毕后,加入吡啶0.39g(3.0wt%);然后升温至25~35℃保温1h;继续升温至80~90℃保温1h;取样检测对氯苯胺残留为0.2%,体系澄清;
[0026]
(3)反应结束后,向反应液中加入2,6
‑
二氟苯甲酰胺16.51g(0.105mol),然后升温至142℃回流反应;保温5h后hplc检测中间体对氯苯基异氰酸酯残留0.3%,反应结束;
[0027]
(4)降温析晶,得除虫脲粗品,粗品水洗,干燥,得除虫脲31.4g,收率97.2%;外观:白色固体;hplc检测含量98%。
[0028]
实施例2
[0029]
(1)取12.11g(0.041mol)三光气置于500ml三口瓶中,加入36.33g氯苯,搅拌溶清,降温至
‑
5~5℃;另取13.00g(0.102mol)对氯苯胺,加入104g氯苯溶清,滴加至三光气溶液中;滴加过程控制反应体系温度为
‑
5~5℃;
[0030]
(2)滴加完毕后,加入三乙胺0.52g(4.0wt%);然后升温至25~35℃保温1h;继续
升温至80~90℃保温1h;取样检测对氯苯胺残留为0.4%,体系澄清;
[0031]
(3)反应结束后,向反应液中加入2,6
‑
二氟苯甲酰胺16.19g(0.103mol),然后升温至130℃回流反应;保温6h后hplc检测中间体对氯苯基异氰酸酯残留0.8%,反应结束;
[0032]
(4)降温析晶,得除虫脲粗品,粗品水洗,干燥,得除虫脲30.9g,收率95.0%;外观:白色固体;hplc检测含量97.5%。
[0033]
实施例3
[0034]
(1)取12.11g(0.041mol)三光气置于500ml三口瓶中,加入48.44g石油醚(沸程90~120℃),搅拌溶清,降温至
‑
5~5℃;另取13.00g(0.102mol)对氯苯胺,加入143g石油醚(沸程90~120℃)溶清,滴加至三光气溶液中;滴加过程控制反应体系温度为
‑
5~5℃;
[0035]
(2)滴加完毕后,加入n’n
‑
二甲基甲酰胺0.65g(5.0wt%);然后升温至25~35℃保温1h;继续升温至80~90℃保温1h;取样检测对氯苯胺残留为0.2%,体系澄清;
[0036]
(3)反应结束后,向反应液中加入2,6
‑
二氟苯甲酰胺16.83g(0.107mol),然后升温至103℃回流反应;保温16h后hplc检测中间体对氯苯基异氰酸酯残留1.0%,反应结束;
[0037]
(4)降温析晶,得除虫脲粗品,粗品水洗,干燥,得除虫脲29.7g,收率91.1%;外观:白色固体;hplc检测含量97.0%。
[0038]
实施例4
[0039]
(1)取12.11g(0.041mol)三光气置于500ml三口瓶中,加入24.2g甲苯,搅拌溶清,降温至
‑
5~5℃;另取13.00g(0.102mol)对氯苯胺,加入117g甲苯溶清,滴加至三光气溶液中;滴加过程控制反应体系温度为
‑
5~5℃;
[0040]
(2)滴加完毕后,加入三苯基膦0.65g(5.0wt%);然后升温至25~35℃保温1h;继续升温至80~90℃保温1h;取样检测对氯苯胺残留为0.5%,体系澄清;
[0041]
(3)反应结束后,向反应液中加入2,6
‑
二氟苯甲酰胺16.83g(0.107mol),然后升温至103℃回流反应;保温16h后hplc检测中间体对氯苯基异氰酸酯残留1.0%,反应结束;
[0042]
(4)降温析晶,得除虫脲粗品,粗品水洗,干燥,得除虫脲29.4g,收率90.1%;外观:白色固体;hplc检测含量97.0%。
[0043]
对比例1
[0044]
(1)取12.11g(0.041mol)三光气置于500ml三口瓶中,加入24.2g二甲苯,搅拌溶清,降温至
‑
5~5℃;另取13.00g(0.102mol)对氯苯胺,加入117g二甲苯溶清,滴加至三光气溶液中;滴加过程控制反应体系温度为
‑
5~5℃;
[0045]
(2)滴加完毕后升温至25~35℃保温1h;继续升温至80~90℃保温1h;取样检测对氯苯胺残留为5%,体系呈固液混合状态,继续80~90℃保温1h后取样检测对氯苯胺残留为0.3%,体系仍呈固液混合状态;
[0046]
(3)趁热过滤除去白色固体杂质约4.3g,滤液经hplc检测,使用面积归一化法计算出滤液中异氰酸酯含量为98%,根据酯化反应情况向滤液中加入2,6
‑
二氟苯甲酰胺12g(0.076mol),然后升温至103℃回流反应;保温16h后hplc检测中间体对氯苯基异氰酸酯残留1.0%,反应结束;
[0047]
(4)降温析晶,得除虫脲粗品,粗品水洗,干燥,得除虫脲17.7g,收率54.1%;外观:白色固体;hplc检测含量96.5%。
[0048]
对比例2
[0049]
(1)取12.11g(0.041mol)三光气置于500ml三口瓶中,加入24.2g二氯乙烷,搅拌溶清,降温至
‑
5~5℃;另取26.00g(0.204mol)对氯苯胺,加入130g二氯乙烷溶清,滴加至三光气溶液中;滴加过程控制反应体系温度为
‑
5~5℃;
[0050]
(2)滴加完毕后,加入n,n
‑
二甲基甲酰胺0.65g(5.0wt%),升温至25~35℃保温1h;继续升温至80~90℃保温1h;取样检测对氯苯胺残留为0.2%,体系澄清;
[0051]
(3)直接降温结晶,析出固体很少,故负压脱出二氯乙烷100g后继续降温结晶,得到中间体异氰酸酯28g,hplc检测含量80.5%;
[0052]
(4)另取一500ml三口瓶,加入上步所得异氰酸酯25g(0.13mol),加入二甲苯100g,加入2,6
‑
二氟苯甲酰胺21.1g(0.134mol),然后升温至139℃回流反应;保温6h后hplc检测中间体对氯苯基异氰酸酯残留1.0%,反应结束;
[0053]
(5)降温析晶,得除虫脲粗品,粗品水洗,干燥,得除虫脲42.1g,两步总收率69.02%;外观:白色固体;hplc检测含量92.0%。
[0054]
通过实施例与对比例1的比较可以看出,本发明在制备异氰酸酯时,未加催化剂时,异氰酸酯反应液中产生了大量的固体杂质,影响了后续反应的收率。通过实施例与对比例2的比较可以看出,在对异氰酸酯进行分离后,所得异氰酸酯的纯度明显下降,这是由于异氰酸酯在分离过程中容易与空气中的水分发生水解反应,在分离中不可避免的有部分中间体发生降解,影响了后续反应的收率。本发明在制备异氰酸酯时,采用的催化剂有效的促进异氰酸酯的生成,抑制杂质的产生。生成的异氰酸酯中间体无需分离提纯,可直接进行下步的合成,节省了分离提纯过程的同时,还降低了异氰酸酯中间体降解的风险。本发明具有工艺简单、副反应少、产品品质高、收率高、操作安全环保等优点,为工业化生产提供良好条件。
[0055]
尽管通过优选实施例的方式对本发明进行了详细描述,但本发明并不限于此。在不脱离本发明的精神和实质的前提下,本领域普通技术人员可以对本发明的实施例进行各种等效的修改或替换,而这些修改或替换都应在本发明的涵盖范围内/任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以权利要求所述的保护范围为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。