一种n
‑
甲基紫杉醇c的制备方法
技术领域
1.本发明涉及紫杉烷类天然产物制备技术领域,尤其涉及一种n
‑
甲基紫杉醇c的制备方法。
背景技术:
2.紫杉醇,一种天然抗癌药物,在临床上已经广泛用于乳腺癌、卵巢癌和部分头颈癌和肺癌的治疗。紫杉醇作为一个具有抗癌活性的二萜生物碱类化合物,其新颖复杂的化学结构、广泛而显著的生物活性、全新独特的作用机制、奇缺的自然资源使其受到了植物学家、化学家、药理学家、分子生物学家的极大青睐,使其成为20世纪下半叶举世瞩目的抗癌明星和研究重点。
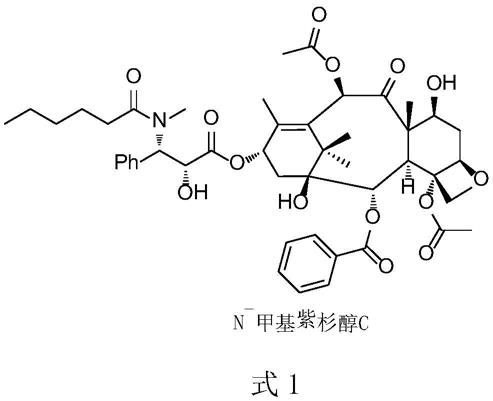
3.作为一个成熟的原料药品种,其杂质谱研究已经非常充分。其中,欧洲药典(ep)收载了一个特征杂质n
‑
甲基紫杉醇c(n
‑
methylpaclitaxel c),结构见式1,为了更好的进行该杂质的研究,制备该杂质变成各原料药厂家必须完成的目标。通过检索发现,基本没有文献报道以可及化合物为原料的合成方法,而从众多天然提取物中进行该杂质的分离又非常困难,因此开发具有可行性的制备方法有非常重要的意义。
4.
技术实现要素:
5.针对上述现有技术的缺点,本发明的目的是提供一种n
‑
甲基紫杉醇c的制备方法,其优点在于起始物料具有可及性,反应条件温和,目标产物纯度高,有利于进行大量制备。
6.本发明的上述技术目的是通过以下技术方案得以实现的:
7.一种n
‑
甲基紫杉醇c的制备方法,包括以下步骤:
8.s1:甲基紫杉醇timf
‑
1以甲酸溶解,室温搅拌反应,反应完成后加入二氯甲烷和饱和碳酸氢钠溶液,收集并浓缩有机相,得timf
‑
2;
9.s2:将timf
‑
2溶于n,n
‑
二甲基甲酰胺中,加入2
‑
甲基咪唑,滴加三乙基氯硅烷,搅拌反应,反应结束后,加水淬灭反应,加乙酸乙酯萃取,浓缩有机相得timf
‑
3;
10.s3:将timf
‑
3溶于二氯甲烷中,加入正己酸和4
‑
二甲氨基吡啶,滴加n,n
‑
二异丙基碳二亚胺,搅拌反应,反应结束后,加水萃取,收集有机相浓缩、过柱得timf
‑
4;
11.s4:将timf
‑
4溶于二氯甲烷中,加入1,8
‑
双二甲氨基萘和三甲基氧鎓四氟硼酸,搅拌反应,反应结束后,将反应液抽滤,滤液加稀盐酸萃取,收集有机相浓缩得timf
‑
5;
12.s5:将timf
‑
5用甲醇和乙酸溶清,加入锌粉,搅拌反应,反应结束后,反应液抽滤,滤液用饱和碳酸氢钠溶液中和,有机相浓缩、过柱得timf
‑
6;
13.s6:将timf
‑
6用n,n
‑
二甲基甲酰胺溶清,加入2
‑
甲基咪唑,滴加三乙基氯硅烷,搅拌反应,反应结束后,加水淬灭反应,加乙酸乙酯萃取,浓缩有机相得timf
‑
7;
14.s7:timf
‑
7用四氢呋喃溶清,加入4
‑
二甲氨基吡啶,滴加乙酸酐,室温搅拌反应,反应结束后,加水淬灭反应,浓缩后抽滤得timf
‑
8;
15.s8:timf
‑
8用乙腈溶清,加入3mol/l盐酸,室温搅拌反应,反应结束后,加二氯甲烷和饱和碳酸氢钠溶液萃取,浓缩有机相过柱、干燥得n
‑
甲基紫杉醇c;
16.上述制备过程发生的反应路线如下:
[0017][0018]
进一步的,在步骤s1中,甲酸与紫杉醇的比例为10(w/v),反应温度范围是0~35℃,反应时间范围是3~4h。
[0019]
进一步的,在步骤s2中,2
‑
甲基咪唑与timf
‑
2的重量比为0.79~0.89,三乙基氯硅烷与与timf
‑
2的重量比为0.15~0.17,反应温度为0~10℃,反应时间范围是0.5~2h。
[0020]
进一步的,在步骤s3中,正己酸与timf
‑
3的重量比为0.124~0.129,二甲氨基吡啶与timf
‑
3的重量比为0.008~0.032,n,n
‑
二异丙基碳二亚胺与timf
‑
3的重量比为0.179~0.242,反应时间范围是1.5~2h。
[0021]
进一步的,在步骤s4中,1,8
‑
双二甲氨基萘与timf
‑
4的重量比为0.5~0.8,三甲基氧鎓四氟硼酸与timf
‑
4的重量比为0.3~0.35,反应温度范围为20~30℃,反应时间范围是4~8h。
[0022]
进一步的,在步骤s5中,锌粉与timf
‑
5的重量比为0.48~0.53,反应时间范围是2~2.5h。
[0023]
进一步的,在步骤s6中,2
‑
甲基咪唑与timf
‑
6的重量比为0.13~0.21,三乙基氯硅烷与timf
‑
6的重量比为0.23~0.29,反应温度为冰浴0℃,反应时间范围是30~45min。
[0024]
进一步的,在步骤s7中,二甲氨基吡啶与timf
‑
7的重量比为0.12~0.18,乙酸酐与timf
‑
7的重量比为0.12~0.18,反应时间为1~3h。
[0025]
进一步的,在步骤s8中,盐酸的浓度为3~3.5mol/l,盐酸与timf
‑
8的比例为1.22~1.33(w/v),二氯甲烷与timf
‑
8的比例为7.31~7.94(w/v)。
[0026]
综上所述,本发明具有以下有益效果:
[0027]
1.起始物料timf
‑
1可以通过10
‑
dab进行3步反应可以轻松制取,相较于一些结构类似的天然提取物杂质,本发明的起始物料更易获取,相应的原料成本更低,有利于大批量的工业生产。
[0028]
2.本发明通过交叉保护,使各步骤主反应明确,基本都没有显著副反应,产物点易于判断;各中间体稳定性较好,特别是裸氨基中间体timf
‑
2不容易发生氧化,timf
‑
2相较于传统制备过程产生的产物具有更好的稳定性,对于整体收率以及后续反应产物判断都有显著提升。
附图说明
[0029]
附图1是实施例1中制备所得n
‑
甲基紫杉醇c的hplc图谱。
[0030]
附图2是实施例1中n
‑
甲基紫杉醇c的ms图谱(正离子模式)。
[0031]
附图3是实施例1中n
‑
甲基紫杉醇c的1h nmr图谱。
[0032]
附图4是实施例1中n
‑
甲基紫杉醇c的
13
c nmr图谱。
具体实施方式
[0033]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图和具体实施方式对本发明提出的装置作进一步详细说明。根据下面说明,本发明的优点和特征将更清楚。为了使本发明的目的、特征和优点能够更加明显易懂,请参阅附图。须知,本说明书所附图式均仅用以配合说明书所揭示的内容,以供熟悉此技术的人士了解与阅读,并非用以限定本发明实施的限定条件,故不具技术上的实质意义,任何结构的修饰、比例关系的改变或大小的调整,在不影响本发明所能产生的功效及所能达成的目的下,均应仍落在本发明所揭示的技术内容能涵盖的范围内。
[0034]
实施例1:
[0035]
s1:将50g紫杉醇timf
‑
1以500ml甲酸溶解,室温(23℃左右)搅拌反应4h,反应完成后加入500ml二氯甲烷,再加饱和碳酸氢钠溶液中和至不冒泡,萃取分液收集并浓缩有机相,得59.4g粘稠物timf
‑
2。
[0036]
s2:将上述timf
‑
2溶于250mln,n
‑
二甲基甲酰胺中,加入5.3g(1.5eq)2
‑
甲基咪唑,滴加9.7g三乙基氯硅烷,冰浴下(0℃)搅拌反应2h,反应结束后,加水淬灭反应,加500ml乙酸乙酯萃取,有机相用纯化水洗2次,浓缩有机相得60.3g粘稠物timf
‑
3。
[0037]
s3:将上述timf
‑
3溶于400ml二氯甲烷中,加入7.5g正己酸和0.5g 4
‑
二甲氨基吡
啶,滴加10.8g n,n
‑
二异丙基碳二亚胺,室温搅拌反应2h,反应结束后,加水萃取,收集有机相浓缩,过柱得42.8g timf
‑
4。
[0038]
s4:将40g timf
‑
4溶于400ml二氯甲烷中,加入20g 1,8
‑
双二甲氨基萘和12g三甲基氧鎓四氟硼酸,30℃搅拌反应4h,反应结束后,将反应液抽滤,滤液加0.1mol/l稀盐酸萃取,收集有机相浓缩得70.3g胶状物timf
‑
5。
[0039]
s5:将上述timf
‑
5用400ml甲醇和120ml乙酸溶清,加入35g锌粉,室温搅拌反应2h,反应结束后,反应液抽滤,滤液用饱和碳酸氢钠溶液中和,有机相浓缩,过柱得25.0g timf
‑
6。
[0040]
s6:25.0g timf
‑
6用125ml n,n
‑
二甲基甲酰胺溶清,加入3.3g 2
‑
甲基咪唑,冰浴下搅拌(0℃),滴加6g三乙基氯硅烷,继续冰浴搅拌反应30min,反应结束后,加水淬灭反应,加乙酸乙酯萃取,浓缩有机相得37.3g胶状物timf
‑
7。
[0041]
s7:将上述timf
‑
7用200ml四氢呋喃溶清,加入6.5g 4
‑
二甲氨基吡啶,滴加5.4g乙酸酐,室温搅拌反应2h。反应结束后,加500ml水淬灭反应,浓缩后抽滤得37.8g timf
‑
8(湿品)。
[0042]
s8:将上述timf
‑
8用250ml乙腈溶清,加入50ml 3mol/l盐酸,室温搅拌反应2h。反应结束后,加300ml二氯甲烷,再加饱和碳酸氢钠溶液至无气泡产生,萃取分液,收集浓缩有机相,再经过过柱、干燥得16.1gn
‑
甲基紫杉醇c,hplc纯度97.7%,总收率为43.3%,液相图谱见附图1。
[0043]
如附图2所示,本实施例中n
‑
甲基紫杉醇c的ms图谱,其中,其质荷比m/z=884.3[m na]
,故其分子式为c
47
h
59
no
14
。
[0044]
如附图3所述,本实施例n
‑
甲基紫杉醇c的1hnmr图谱,其中,1h nmr(500mhz,cdcl3)δ:7.33~8.11(10h,arh),6.30(1h,s,h
10
),6.17(1h,t,j=8.6hz,h
13
),5.78(1h,d,j=2.9hz,h
32
),5.68(1h,d,j=7.0hz,h2),4.93(1h,d,j=8.8hz,h5),4.89(1h,d,j=3.3hz,h
31
),4.41(1h,dd,j=10.7hz,6.8hz,h7),4.28(1h,d,j=8.4hz,h
20
),4.17(1h,d,j=8.4hz,h
20
),3.80(1h,d,j=6.9hz,h3),2.89(3h,s,h
38
),2.55(1h,m,h6),2.34(4h,m,2h
14 overlapped with 2h
40
),2.24(3h,s,h
27
),2.22(3h,s,h
29
),1.89(3h,s,h
18
),1.86(3h,m,2
×‑
oh overlapped with h6),1.67(3h,s,h
19
),1.60(2h,m,h
41
),1.28(7h,m,2h
42
/2h
43 overlapped with 3h
44
),1.15(3h,s,h
16
/h
17
),0.86(3h,s,h
16
/h
17
)。碳原子编号参考以下结构式:
[0045]
[0046]
如附图4所示,本实施例n
‑
甲基紫杉醇c
13
c nmr图谱,其中,13c nmr(125mhz,cdcl3)δ:203.853,175.054,173.588,171.223,170.213,166.948,142.752,136.655,133.616,132.843,130.212,129.298,128.881,128.636,128.131,128.098,84.446,80.9,79.124,76.457,75.681,75.119,72.839,72.129,61.509,58.593,45.627,43.237,35.728,35.574,35.337,34.025,31.493,26.759,24.704,22.385,22.194,21.924,20.812,14.796,13.853,9.544。
[0047]
实施例2:
[0048]
s1:将5g timf
‑
1以50ml甲酸溶解,室温(25℃左右)搅拌反应3h,反应完成后加入50ml二氯甲烷,再加饱和碳酸氢钠溶液中和至不冒泡,萃取分液收集并浓缩有机相,得6.0g粘稠物timf
‑
2。
[0049]
s2:将上述timf
‑
2溶于25mln,n
‑
二甲基甲酰胺中,加入0.5g 2
‑
甲基咪唑,滴加1g三乙基氯硅烷,10℃以下搅拌反应30min,反应结束后,加水淬灭反应,加50ml乙酸乙酯萃取,有机相用纯化水洗2次,浓缩有机相得6.0g粘稠物timf
‑
3;
[0050]
s3:将上述timf
‑
3溶于40ml二氯甲烷中,加入0.75g正己酸和0.1g 4
‑
二甲氨基吡啶,滴加1.1g n,n
‑
二异丙基碳二亚胺,室温搅拌反应2h,反应结束后,加水萃取,收集有机相浓缩,过柱得4.3g timf
‑
4。
[0051]
s4:将4.3g timf
‑
4溶于40ml二氯甲烷中,加入3.4g 1,8
‑
双二甲氨基萘和1.5g三甲基氧鎓四氟硼酸,20℃搅拌反应8h,反应结束后,将反应液抽滤,滤液加0.1mol/l稀盐酸萃取,收集有机相浓缩得7.2g胶状物timf
‑
5。
[0052]
s5:将上述timf
‑
5用40ml甲醇和12ml乙酸溶清,加入3.5g锌粉,室温搅拌反应2h,反应结束后,反应液抽滤,滤液用饱和碳酸氢钠溶液中和,有机相浓缩,过柱得2.6g timf
‑
6。
[0053]
s6:2.6g timf
‑
6用15ml n,n
‑
二甲基甲酰胺溶清,加入0.5g 2
‑
甲基咪唑,冰浴下搅拌(0℃),滴加0.6g三乙基氯硅烷,继续冰浴搅拌反应30min,反应结束后,加水淬灭反应,加乙酸乙酯萃取,浓缩有机相3.8g得胶状物timf
‑
7。
[0054]
s7:将上述timf
‑
7用20ml四氢呋喃溶清,加入0.6g 4
‑
二甲氨基吡啶,滴加0.5g乙酸酐,室温搅拌反应2h。反应结束后,加80ml水淬灭反应,浓缩后抽滤得3.8g timf
‑
8(湿品)。
[0055]
s8:将上述timf
‑
8用25ml乙腈溶清,加入5ml 3mol/l盐酸,室温搅拌反应2h。反应结束后,加二氯甲烷,再加饱和碳酸氢钠溶液至无气泡产生,萃取分液,收集浓缩有机相,再经过过柱、干燥得1.8g n
‑
甲基紫杉醇c,hplc纯度98.8%,总收率为48.4%。
[0056]
实施例3:
[0057]
s1:将5g timf
‑
1以50ml甲酸溶解,室温(25℃左右)搅拌反应3h,反应完成后加入50ml二氯甲烷,再加饱和碳酸氢钠溶液中和至不冒泡,萃取分液收集并浓缩有机相,得6.3g粘稠物timf
‑
2。
[0058]
s2:将上述timf
‑
2溶于25mln,n
‑
二甲基甲酰胺中,加入0.5g 2
‑
甲基咪唑,滴加1g三乙基氯硅烷,5℃左右搅拌反应45min,反应结束后,加水淬灭反应,加50ml乙酸乙酯萃取,有机相用纯化水洗2次,浓缩有机相得6.2g粘稠物timf
‑
3;
[0059]
s3:将上述timf
‑
3溶于40ml二氯甲烷中,加入0.8g正己酸和0.2g 4
‑
二甲氨基吡
啶,滴加1.5g n,n
‑
二异丙基碳二亚胺,室温搅拌反应1.5h,反应结束后,加水萃取,收集有机相浓缩,过柱得4.7g timf
‑
4。
[0060]
s4:将4.7g timf
‑
4溶于90ml二氯甲烷中,加入3.5g 1,8
‑
双二甲氨基萘和1.5g三甲基氧鎓四氟硼酸,室温26℃左右搅拌反应6h,反应结束后,将反应液抽滤,滤液加0.1mol/l稀盐酸萃取,收集有机相浓缩得7.5g胶状物timf
‑
5。
[0061]
s5:将上述timf
‑
5用50ml甲醇和15ml乙酸溶清,加入4.0g锌粉,室温搅拌反应2.5h,反应结束后,反应液抽滤,滤液用饱和碳酸氢钠溶液中和,有机相浓缩,过柱得2.8g timf
‑
6。
[0062]
s6:2.8g timf
‑
6用15ml n,n
‑
二甲基甲酰胺溶清,加入0.6g 2
‑
甲基咪唑,冰浴下搅拌(0℃),滴加0.8g三乙基氯硅烷,继续冰浴搅拌反应45min,反应结束后,加水淬灭反应,加乙酸乙酯萃取,浓缩有机相得4.0g胶状物timf
‑
7。
[0063]
s7:将上述timf
‑
7用20ml四氢呋喃溶清,加入0.8g 4
‑
二甲氨基吡啶,滴加0.8g乙酸酐,室温搅拌反应2h。反应结束后,加80ml水淬灭反应,浓缩后抽滤得timf
‑
8(湿品)。
[0064]
s8:将上述timf
‑
8用25ml乙腈溶清,加入5ml 3mol/l盐酸,室温搅拌反应3h。反应结束后,加二氯甲烷,再加饱和碳酸氢钠溶液至无气泡产生,萃取分液,收集浓缩有机相,再经过过柱、干燥得2.0g n
‑
甲基紫杉醇c,hplc纯度97.5%,总收率为53.7%。
[0065]
对比例1:
[0066]
尝试以紫杉醇timf
‑1’
为原料进行n
‑
甲基紫杉醇c的制备。
[0067]
称取5.0g timf
‑1’
于反应瓶中,加入50ml甲酸,搅拌溶清,室温搅拌反应3h,反应完成后加入50ml二氯甲烷,再加饱和碳酸氢钠溶液中和至不冒泡,萃取分液收集并浓缩有机相,得粘稠物timf
‑2’
,tlc监测产物为两个极性较为相近的点,比例接近1:1;
[0068]
s2:将上述timf
‑2’
溶于25mln,n
‑
二甲基甲酰胺中,加入0.5g 2
‑
甲基咪唑,滴加1g三乙基氯硅烷,10℃以下搅拌反应30min,反应结束后,加水淬灭反应,加50ml乙酸乙酯萃取,有机相用纯化水洗2次,浓缩有机相得粘稠物,此时的产物较为复杂,无法分辨哪个是主产物timf
‑3’
,放弃此路线;
[0069][0070]
结论:timf
‑2’
的氨基容易被氧化,导致生成降解产物,继而在步骤2中生产对应的更多种类产物,该路线整体的收率有显著降低,而且无法简单识别哪个是目标结构的中间体,同时也提高了纯化难度。
[0071]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0072]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。