抗肿瘤药cfi
‑
402257的合成制备工艺
技术领域
1.本发明涉及生物医药技术领域,具体地,涉及一种抗肿瘤药物cfi
‑
402257的合成制备工艺。
背景技术:
2.treadwell治疗公司获得了university healthnetwork(加拿大安大略省大学健康网络)的许可,开发一种有效的、高度选择性和口服活性、作为单极纺锤体蛋白激酶1(mps1)小分子抑制剂cfi
‑
402257口服胶囊配方。cfi
‑
402257目前用于转移性乳腺癌(metastatic breast cancer)的适应症在加拿大处于2期临床,用于晚期实体瘤(advanced solid tumor)的适应症在加拿大处于1期临床。
3.用cfi
‑
402257处理的人癌细胞表现出与mps1激酶抑制相一致的作用,特别是cfi
‑
402257治疗会导致sac失活、染色体失配、非整倍体,最终导致细胞死亡。cfi
‑
402257在肿瘤细胞系和患者源性异种移植(pdx)模型中显示了单药治疗的有效性。在高级别浆液性卵巢癌的铂耐药pdx模型中也显示了抗肿瘤活性。cfi
‑
402257诱导基因组不稳定和凋亡细胞死亡,从而促进肿瘤免疫。在小鼠人类肿瘤模型中,单药口服cfi
‑
402257或与抗程序性细胞死亡1(pd
‑
1)抗体联合使用可在耐受性良好的剂量下抑制肿瘤生长。cfi
‑
402257是一种潜在的治疗癌症的小分子抑制剂,其作用机制与mps1的强选择性抑制作用一致(liu y,et al.acs med chem lett,2016,7(7):671
‑
675;jacqueline m,et al.pnas,2017,114(12):3127
‑
3132)。此外,mps1和细胞周期蛋白依赖性激酶4/6抑制的联合应用被认为是增加mps1抑制剂治疗窗口的策略(plos one,2015,10(9):e0138616)。cdk4/6抑制剂可使rb1细胞周期阻滞,从而提高mps1抑制剂的耐受性和疗效。
4.cfi
‑
402257的结构在其化合物专利wo2015070349a1及晶型专利wo2018014116a1中披露,cfi
‑
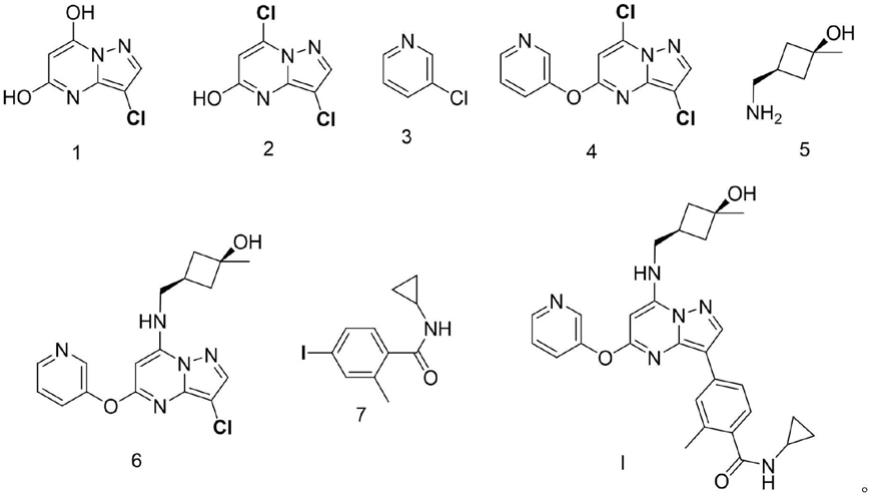
402257的结构式如式i所示化合物:
[0005][0006]
目前cfi
‑
402257的合成制备工艺仍有待改进。
技术实现要素:
[0007]
本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的
一个目的在于提出一种新的合成制备cfi
‑
402257的工艺。相对于现有技术合成路线中在第三步利用boc对羟基进行保护再反应,本技术所述的合成路线无需进行保护,减小一步反应。另外,本技术所述的制备工艺中,最后一步利用有机锌试剂参与的特异性卤代反应,避免了使用化学碱时对反应物中的羟基造成副反应,路线的总体产率更高。
[0008]
在本发明的一个方面,本发明提供了一种式i所示化合物cfi
‑
402257的合成制备工艺。根据本发明的实施例,该方法包括:
[0009]
(1)使式1所示化合物与pocl3接触,以便获得式2所示化合物;
[0010]
(2)使式2所示化合物与式3所示化合物接触,以便获得式4所示化合物;
[0011]
(3)使式4所示化合物与式5所示化合物接触,以便获得式6所示化合物;
[0012]
(4)使式6所示化合物与式7所示化合物接触,以便获得式i所示化合物,
[0013][0014]
发明人发现,利用本发明的该方法,采用已有商业容易获取的中间体化合物,其经过4步化学反应,能够快速、有效地制备获得式i所示化合物。
[0015]
在本文中所使用的术语“接触”应做广义理解,其可以是任何能够使得至少两种反应物发生化学反应的方式,例如可以是将两种反应物在适当的条件下进行混合。根据需要,可以在搅拌下,将需要进行接触的反应物进行混合,由此,搅拌的类型并不受特别限制,例如可以为机械搅拌,即在机械力的作用下进行搅拌。
[0016]
在本文中,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。在本发明的描述中,“多个”的含义是两个或两个以上,除非另有明确具体的限定。
[0017]
根据本发明的实施例,上述制备式2所示化合物、式4所示化合物、式6所示化合物、式i所示化合物的方法还可以具有下列附加技术特征至少之一:
[0018]
根据本发明的实施例,本发明所述的化学反应可以按照本领域已知的任何方法进行。制备式2所示化合物、式4所示化合物、式6所示化合物、式i所示化合物的原料的来源并不受特别限制,其可以是采用任何已知的方法制备的,或者市售获得的。
[0019]
根据本发明的实施例,在步骤(1)中,式1所示化合物与pocl3的接触方式并不受特别限制。由此,可以提升式1所示化合物与pocl3接触反应的效率,加快反应速度,进一步提高利用该方法制备式2所示化合物的效率。
[0020]
根据本发明的实施例,在步骤(1)中,包括如下步骤:将式1所示化合物加入乙腈中搅拌,缓慢加入pocl3,将混合物在室温搅拌接触反应,反应毕,将混合物倒在冰上,用固体碳酸氢钠中和至ph值约为8,然后用乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用柱层析纯化得式2所示化合物。由此,可以提升式1所示化合物与pocl3接触反应的效率,加快反应速度,进一步提高利用该方法制备式2所示化合物的效率。
[0021]
根据本发明的实施例,在步骤(1)中,式1所示化合物与pocl3的摩尔比为1:(0.7~0.9),优选式1所示化合物与pocl3的摩尔比为1:0.75。由此,反应物利用率较高,不会造成原料、实际的浪费,目标化合物收率较高,可以进一步提高利用该方法制备式2所示化合物的效率。
[0022]
根据本发明的实施例,在步骤(1)中,式1所示化合物与pocl3搅拌接触的反应时间为10~15h,优选反应时间为12小时,由此,可以提升式1所示化合物与pocl3接触的效率,进一步提高利用该方法制备式2所示化合物的效率。
[0023]
根据本发明的实施例,在步骤(1)中,所述柱层析采用体积比为(8~20):1的石油醚/乙酸乙酯的混合溶剂,优选柱层析采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂。
[0024]
根据本发明的一个具体实施例,在步骤(1)中,包括如下步骤:将5.0g式1所示化合物加入30ml乙腈中搅拌,缓慢加入3.1g pocl3,将混合物在室温搅拌接触反应12小时,反应毕,将混合物倒在冰上,用固体碳酸氢钠中和至ph值约为8,然后用3
×
30ml乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式2所示化合物,得量4.8g,收率87.3%。
[0025]
根据本发明的实施例,在步骤(2)中,式2所示化合物、式3所示化合物与k2co3的接触方式并不受特别限制。由此,可以提升式2所示化合物、式3所示化合物与k2co3接触反应的效率,加快反应速度,进一步提高利用该方法制备式4所示化合物的效率。
[0026]
根据本发明的实施例,在步骤(2)中,包括如下步骤:在ch2cl2中加入式2所示化合物和式3所示化合物,搅拌,加入k2co3,将混合物在室温搅拌接触反应,反应毕,将混合物倒入冰水中洗涤,用乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后柱层析纯化得式4所示化合物。由此,可以提升式2所示化合物、式3所示化合物与k2co3接触反应的效率,加快反应速度,进一步提高利用该方法制备式4所示化合物的效率。
[0027]
根据本发明的实施例,在步骤(2)中,式2所示化合物、式3所示化合物、k2co3的摩尔比为1:(1.0~1.2):(1.1~1.5),优选式2所示化合物、式3所示化合物、k2co3的摩尔比为1:1:1.2。由此,可以进一步提高利用该方法制备式4所示化合物的效率。
[0028]
根据本发明的实施例,在步骤(2)中,式2所示化合物、式3所示化合物、k2co3接触搅拌的反应时间为10~15h,优选式2所示化合物、式3所示化合物、k2co3搅拌接触的反应时间为12小时。由此,可以提升式2所示化合物、式3所示化合物、k2co3接触反应的效率,进一步提高利用该方法制备式4所示化合物的效率。
[0029]
根据本发明的实施例,在步骤(2)中,所述柱层析采用体积比为(8~20):1的石油醚/乙酸乙酯的混合溶剂,优选柱层析采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂。
[0030]
根据本发明的一个具体实施例,在步骤(2)中,包括如下步骤:在45ml ch2cl2中加入4.50g式2所示化合物和2.50g式3所示化合物,搅拌,加入3.66g k2co3,将混合物在室温搅拌接触反应12小时,反应毕,将混合物倒入45ml冰水中洗涤,用3
×
45ml乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式4所示化合物,得量5.6g,收率90.3%。
[0031]
根据本发明的实施例,在步骤(3)中,式4所示化合物、式5所示化合物、三乙胺的接触方式并不受特别限制。由此,可以提升式4所示化合物、式5所示化合物、三乙胺接触反应的效率,加快反应速度,进一步提高利用该方法制备式6所示化合物的效率。
[0032]
根据本发明的实施例,在步骤(3)中,包括如下步骤:在mecn中加入式4所示化合物和式5所示化合物,搅拌,加入三乙胺,将混合物在室温搅拌反应,反应毕,将混合物倒入冰水中洗涤,用乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后柱层析纯化得式6所示化合物。由此,可以进一步提高利用该方法制备式5所示化合物的效率。
[0033]
根据本发明的实施例,在步骤(3)中,式4所示化合物、式5所示化合物、三乙胺的摩尔比为1:(1.0~1.2):(1.1~1.5),优选式4所示化合物、式5所示化合物、三乙胺的摩尔比为1:1.05:1.2。由此,反应物利用率较高,不会造成原料、实际的浪费,目标化合物收率较高。
[0034]
根据本发明的实施例,在步骤(3)中,式4所示化合物、式5所示化合物、三乙胺接触搅拌的反应时间为10~15h,优选式4所示化合物、式5所示化合物、三乙胺搅拌接触的反应时间为12小时。由此,可以提升式4所示化合物、式5所示化合物、三乙胺接触反应的效率,进一步提高利用该方法制备式6所示化合物的效率。
[0035]
根据本发明的实施例,在步骤(3)中,所述柱层析采用体积比为(8~20):1的石油醚/乙酸乙酯的混合溶剂,优选柱层析采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂。
[0036]
根据本发明的一个具体实施例,在步骤(3)中,包括如下步骤:在40mlmecn中加入4.0g式4所示化合物和1.72g式5所示化合物,搅拌,加入1.73g三乙胺,将混合物在室温搅拌反应12小时,反应毕,将混合物倒入45ml冰水中洗涤,用3
×
40ml乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式6所示化合物,得量4.50g,收率87.9%。
[0037]
根据本发明的实施例,在步骤(4)中,式7所示化合物、rieke锌、式6所示化合物、pdcl2(dppf)的接触方式并不受特别限制。由此,可以提升式7所示化合物、rieke锌、式6所示化合物、pdcl2(dppf)接触反应的效率,加快反应速度,进一步提高利用该方法制备式i所示化合物的效率。
[0038]
根据本发明的实施例,在步骤(4)中,包括如下步骤:n2保护下,将式7所示化合物溶解于dmf中并由注射器快速加入rieke锌,在25℃下搅拌15min,然后将容器在n2气流下打开,依次加入式6所示化合物和pdcl2(dppf),将反应混合物升温搅拌反应,反应毕,将反应混合物冷却至室温后加入ea稀释,经硅藻土过滤,向滤液中加h2o,有机相用盐水洗涤,硫酸钠干燥,过滤,滤液浓缩后柱层析纯化得式i所示化合物。由此,可以进一步提高利用该方法制备式i所示化合物的效率。
[0039]
根据本发明的实施例,在步骤(4)中,式7所示化合物、rieke锌、式6所示化合物、pdcl2(dppf)的摩尔比为(1.05~1.2):(1.0~1.3):1:(0.025~0.1),优选式7所示化合物、
rieke锌、式6所示化合物、pdcl2(dppf)的摩尔比为1.1:1.1:1:0.05。由此,反应物利用率较高,不会造成原料、实际的浪费,目标化合物收率较高。
[0040]
根据本发明的实施例,在步骤(4)中,式7所示化合物、rieke锌、式6所示化合物、pdcl2(dppf)接触搅拌的反应时间为45分钟~90分钟,优选式7所示化合物、rieke锌、式6所示化合物、pdcl2(dppf)接触搅拌的反应时间为1小时。由此,可以提升式7所示化合物、rieke锌、式6所示化合物、pdcl2(dppf)接触反应的效率,进一步提高利用该方法制备式i所示化合物的效率。
[0041]
根据本发明的实施例,在步骤(4)中,式7所示化合物、rieke锌、式6所示化合物、pdcl2(dppf)接触搅拌的反应温度为70~85℃,优选式7所示化合物、rieke锌、式6所示化合物、pdcl2(dppf)接触搅拌的反应温度为80℃。由此,可以提升式7所示化合物、rieke锌、式6所示化合物、pdcl2(dppf)接触反应的效率,进一步提高利用该方法制备式i所示化合物的效率。
[0042]
根据本发明的实施例,在步骤(4)中,所述柱层析采用体积比为(8~20):1的石油醚/乙酸乙酯的混合溶剂,优选柱层析采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂。
[0043]
根据本发明的一个具体实施例,在步骤(4)中,包括如下步骤:n2保护下,将3.5g式7所示化合物溶解于35ml dmf中并由注射器快速加入已配制的含0.76g rieke锌的50mg/ml的rieke锌
‑
thf混合液,加毕,在25℃下搅拌15min,然后将容器在n2气流下打开,依次加入3.8g式6所示化合物和386mg pdcl2(dppf),将反应混合物升温至80℃反应1小时,反应毕,将反应混合物冷却至室温后加入35ml乙酸乙酯稀释,经硅藻土过滤,向滤液中加35ml水,有机相用盐水洗涤,硫酸钠干燥,过滤,滤液浓缩后采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式i所示化合物,得量4.46g,收率84.7%,hplc纯度99.7%。
[0044]
根据本发明的实施例,式i所示化合物cfi
‑
402257的合成路线可以如下所示:
[0045][0046]
相对于现有技术,本发明所述抗肿瘤药cfi
‑
402257的制备工艺,其至少具有以下有益效果:
[0047]
1、本发明所述的方法,采用已有商业容易获取的中间体化合物,其经过共4步反应
合成目标分子cfi
‑
402257。
[0048]
2、本发明所述的合成方法,其在步骤(1)中采用pocl3特异性氯化式1所示化合物中的7位上的羟基,避免了式1所示化合物中的两个
‑
oh均参与与式3所示化合物进行卤代反应的可能性,其有效提高反应的选择性,以及提高了步骤(2)中卤代反应的产率。
[0049]
3、本发明所述的合成方法,在步骤(3)采用弱碱et3n可以特异性的利用式4所示化合物中的7位上的cl进行卤代反应,提高了步骤(3)反应的选择性。
[0050]
4、相对于现有技术合成路线中在第三步利用boc对羟基进行保护再反应,本技术所述的合成路线在前3步反应中均无需对化合物进行保护,相比于相应技术,本技术的反应过程减少一步反应。
[0051]
5、最后一步利用有机锌试剂,特异性进行卤代反应,避免使用化学碱时式6所示化合物中的
‑
oh与式7所示化合物中的
‑
i(碘)结合而产生副反应。
[0052]
6、相对于现有合成路线的反应步骤,本技术所述的制备工艺路线反应步数更少,各步骤反应难度小,路线的总体产率更高,每步反应产率高并且操作简便,有效提高了反应的总收率和工业化操作性。
具体实施方式
[0053]
下面详细描述本发明的实施例。下面描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0054]
实施例1式2所示化合物的合成
[0055]
将式1所示化合物(5.0g,26.94mmol)加入乙腈(30ml)中搅拌,缓慢加入pocl3(3.1g,20.21mmol),将混合物在室温搅拌接触反应12小时,反应毕,将混合物倒在冰上,用固体碳酸氢钠中和至ph值约为8,然后用乙酸乙酯(3
×
30ml)萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式2所示化合物,得量4.8g,收率87.3%。
[0056]
lc
‑
ms(apci):m/z=204.0(m 1)
。
[0057]
实施例2式2所示化合物的合成
[0058]
将式1所示化合物(5.0g,26.94mmol)加入乙腈(30ml)中搅拌,缓慢加入pocl3(2.57g,16.76mmol),将混合物在室温搅拌接触反应10小时,反应毕,将混合物倒在冰上,用固体碳酸氢钠中和至ph值约为8,然后用乙酸乙酯(3
×
30ml)萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为8:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式2所示化合物,得量4.5g,收率81.9%。
[0059]
实施例3式2所示化合物的合成
[0060]
将式1所示化合物(5.0g,26.94mmol)加入乙腈(30ml)中搅拌,缓慢加入pocl3(3.72g,24.25mmol),将混合物在室温搅拌接触反应15小时,反应毕,将混合物倒在冰上,用固体碳酸氢钠中和至ph值约为8,然后用乙酸乙酯(3
×
30ml)萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为20:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式2所示化合物,得量4.7g,收率85.5%。
[0061]
实施例4式4所示化合物的合成
[0062]
在ch2cl2(45ml)中加入式2所示化合物(4.50g,22.06mmol)和式3所示化合物(2.50g,22.06mmol),搅拌,加入k2co3(3.66g,26.47mmol),将混合物在室温搅拌接触反应12小时,反应毕,将混合物倒入冰水(45ml)中洗涤,用乙酸乙酯(3
×
45ml)萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式4所示化合物,得量5.6g,收率90.3%。
[0063]
lc
‑
ms(apci):m/z=282.0(m 1)
。
[0064]
实施例5式4所示化合物的合成
[0065]
在ch2cl2(45ml)中加入式2所示化合物(4.50g,22.06mmol)和式3所示化合物(2.76g,24.31mmol),搅拌,加入k2co3(3.35g,24.24mmol),将混合物在室温搅拌接触反应10小时,反应毕,将混合物倒入冰水(45ml)中洗涤,用乙酸乙酯(3
×
45ml)萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为8:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式4所示化合物,得量5.3g,收率85.5%。
[0066]
实施例6式4所示化合物的合成
[0067]
在ch2cl2(45ml)中加入式2所示化合物(4.50g,22.06mmol)和式3所示化合物(2.55g,22.45mmol),搅拌,加入k2co3(4.57g,33.09mmol),将混合物在室温搅拌接触反应15小时,反应毕,将混合物倒入冰水(45ml)中洗涤,用乙酸乙酯(3
×
45ml)萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为20:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式4所示化合物,得量5.5g,收率88.7%。
[0068]
实施例7式6所示化合物的合成
[0069]
在mecn(40ml)中加入式4所示化合物(4.0g,14.23mmol)和式5所示化合物(1.72g,14.95mmol),搅拌,加入三乙胺(1.73g,17.08mmol),将混合物在室温搅拌反应12小时,反应毕,将混合物倒入冰水中(45ml)洗涤,用乙酸乙酯(3
×
40ml)萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式6所示化合物,得量4.50g,为收率87.9%。
[0070]
lc
‑
ms(apci):m/z=360.2(m 1)
。
[0071]
实施例8式6所示化合物的合成
[0072]
在mecn(40ml)中加入式4所示化合物(4.0g,14.23mmol)和式5所示化合物(1.64g,14.23mmol),搅拌,加入三乙胺(1.79g,15.65mmol),将混合物在室温搅拌反应10小时,反应毕,将混合物倒入冰水中(45ml)洗涤,用乙酸乙酯(3
×
40ml)萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为8:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式6所示化合物,得量4.32g,为收率84.3%。
[0073]
实施例9式6所示化合物的合成
[0074]
在mecn(40ml)中加入式4所示化合物(4.0g,14.23mmol)和式5所示化合物(1.97g,17.10mmol),搅拌,加入三乙胺(2.16g,21.35mmol),将混合物在室温搅拌反应15小时,反应毕,将混合物倒入冰水中(45ml)洗涤,用乙酸乙酯(3
×
40ml)萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩后采用体积比为20:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式6所示化合物,得量4.43g,为收率86.5%。
[0075]
实施例10式i所示化合物的合成
[0076]
n2保护下,将式7所示化合物(3.5g,11.62mmol)溶解于dmf(35ml)中并由注射器快速加入已配制的含0.76g rieke锌(11.62mmol)的50mg/ml的rieke锌
‑
thf混合液,加毕,在25℃下搅拌15min,然后将容器在n2气流下打开,依次加入式6所示化合物(3.8g,10.56mmol)和pdcl2(dppf)(386mg,0.528mmol),将反应混合物升温至80℃反应1小时,反应毕,将反应混合物冷却至室温后加入ea(35ml)稀释,经硅藻土过滤,向滤液中加h2o(35ml),有机相用盐水洗涤,硫酸钠干燥,过滤,滤液浓缩后采用体积比为10:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式i所示化合物,得量4.46g,收率84.7%,hplc纯度99.7%。
[0077]
lc
‑
ms(apci):m/z=499.3(m 1)
。
[0078]
实施例11式i所示化合物的合成
[0079]
n2保护下,将式7所示化合物(3.34g,11.09mmol)溶解于dmf(35ml)中并由注射器快速加入含0.69g rieke锌(10.56mmol)的50mg/ml的rieke锌
‑
thf混合液,加毕,在25℃下搅拌15min,然后将容器在n2气流下打开,依次加入式6所示化合物(3.80g,10.56mmol)和pdcl2(dppf)(193mg,0.264mmol),将反应混合物升温至70℃反应90分钟,反应毕,将反应混合物冷却至室温后加入ea(35ml)稀释,经硅藻土过滤,向滤液中加h2o(35ml),有机相用盐水洗涤,硫酸钠干燥,过滤,滤液浓缩后采用体积比为8:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式i所示化合物,得量3.70g,收率76.3%,hplc纯度99.8%。
[0080]
实施例12式i所示化合物的合成
[0081]
n2保护下,将式7所示化合物(3.82g,12.68mmol)溶解于dmf(35ml)中并由注射器快速加入含0.90g rieke锌(13.73mmol)的50mg/ml的rieke锌
‑
thf混合液,加毕,在25℃下搅拌15min,然后将容器在n2气流下打开,依次加入式6所示化合物(3.80g,10.56mmol)和pdcl2(dppf)(772mg,1.056mmol),将反应混合物升温至85℃反应45分钟,反应毕,将反应混合物冷却至室温后加入ea(35ml)稀释,经硅藻土过滤,向滤液中加h2o(35ml),有机相用盐水洗涤,硫酸钠干燥,过滤,滤液浓缩后采用体积比为20:1的石油醚/乙酸乙酯的混合溶剂柱层析纯化得式i所示化合物,得量4.33g,收率82.3%,hplc纯度99.5%。
[0082]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
[0083]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。