1.本发明属于电解液添加剂合成技术领域,涉及一种3,4-二丙腈-1,1-二氧四氢噻吩的合成方法。
背景技术:
2.电解液添加剂是指为改善电解液的电化学性能和提高阴极沉积质量而加入电解液中的少量添加物。电解液添加剂是一些天然或人工合成的有机和无机化合物,一般不参加电解过程,电解液添加剂用量一般很小,但却是电解质体系不可缺少的部分。3,4-二丙腈-1,1-二氧四氢噻吩是一种噻吩类化合物,应用于电解液添加剂中,能够在正极表面形成薄膜,覆盖正极的活性位点,达到保护正极的目的,防止正极与电解液反应产气,从而改善锂离子电池的循环性能。但是现有的3,4-二丙腈-1,1-二氧四氢噻吩的合成方法存在收率低的问题,从而电解液添加剂中的成本提高。
技术实现要素:
3.本发明提出一种3,4-二丙腈-1,1-二氧四氢噻吩的合成方法,解决了现有技术中3,4-二丙腈-1,1-二氧四氢噻吩的合成方法收率低的问题。
4.本发明的技术方案是这样实现的:
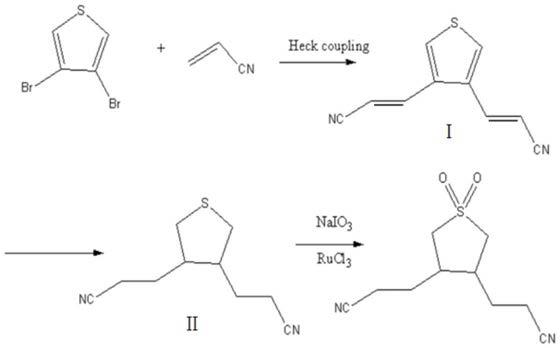
5.一种3,4-二丙腈-1,1-二氧四氢噻吩的合成方法,以3,4-二溴噻吩为原料,与丙烯腈发生heck偶联反应,得到化合物ⅰ,再以化合物ⅰ为原料,催化加氢,得到化合物ⅱ,化合物ⅱ在三氯化钌的催化下,经高碘酸钠氧化反应,得到3,4-二丙腈-1,1-二氧四氢噻吩,
6.其合成路线如下:
[0007][0008]
作为进一步的技术方案,包括以下步骤:
[0009]
a、将3,4-二溴噻吩与乙腈混合,加入吡啶,通入氮气,加入催化剂,升温至50~70℃,加入丙烯腈,反应后得到化合物ⅰ;
[0010]
b、将化合物ⅰ与催化剂混合后,加入乙醇,升温至180~200℃,通入氢气,反应后得到化合物ⅱ;
[0011]
c、将化合物ⅱ溶于乙腈中,在0~5℃加入三氯化钌和高碘酸钠混合物,反应后得
到3,4-二丙腈-1,1-二氧四氢噻吩。
[0012]
作为进一步的技术方案,步骤a中反应时间为2~2.5h,步骤b中反应时间为6~9h,步骤c中反应时间为30~40min。
[0013]
作为进一步的技术方案,步骤a中催化剂由质量比为(8-10):1的双(三苯基膦)氯化镍与锌组成。
[0014]
作为进一步的技术方案,步骤a中催化剂与3,4-二溴噻吩的质量比为(0.01~0.02):1,步骤b中催化剂与化合物ⅰ的质量比为(0.02~0.05):1,步骤c中三氯化钌、高碘酸钠与化合物ⅱ的质量比为3:(0.002~0.006):1。
[0015]
作为进一步的技术方案,步骤a中3,4-二溴噻吩、丙烯腈与吡啶的摩尔比为1:(2.2~2.6):(2.2~2.6)。
[0016]
作为进一步的技术方案,步骤a乙腈与3,4-二溴噻吩的体积质量比为(3~4)ml:1g,步骤b中甲醇与化合物ⅰ的体积质量比为(9~12)ml:1g,步骤c中乙腈与化合物ⅱ的体积质量比为(3~4)ml:1g。
[0017]
作为进一步的技术方案,步骤c中,在0~5℃加入三氯化钌和高碘酸钠混合物前,先降温至5~10℃加入碳酸氢钠水溶液调节ph值至6.8~7.3,三氯化钌和高碘酸钠混合物的加入方式采用滴加,2~3h滴毕。
[0018]
作为进一步的技术方案,步骤a中丙烯腈的加入方式为滴加,2~2.5h滴毕。
[0019]
作为进一步的技术方案,步骤b中催化剂由质量比为(1.5~3)的第一催化剂与第二催化剂组成,所述第一催化剂为钯碳催化剂,所述第二催化剂由质量比为10:7:25的氧化镍、氧化钼及氧化铝载体组成,所述第二催化剂的比表面积为150~200m2/g,孔容为0.5ml/g。
[0020]
本发明的工作原理及有益效果为:
[0021]
1、本发明中,以3,4-二溴噻吩为起始原料,依次经过heck偶联反应、催化加氢反应、氧化反应,得到最终产物3,4-二丙腈-1,1-二氧四氢噻吩,通过对heck偶联反应、催化加氢反应中的催化剂进行优化设计,显著提高了heck偶联反应、催化加氢反应的收率,从而将最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率提高至78.5%以上,有效解决了现有技术中3,4-二丙腈-1,1-二氧四氢噻吩的合成方法收率低的问题。
[0022]
2、本发明中,以3,4-二溴噻吩为起始原料,在催化剂作用下,与丙烯腈发生heck偶联反应,得到化合物ⅰ,采用质量比为(8-10):1的双(三苯基膦)氯化镍与锌混合作为催化剂,特定质量比的双(三苯基膦)氯化镍与锌协同作用,显著提高了化合物i的收率,从而提高最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率。
[0023]
3、本发明中,化合物ⅰ在催化剂的作用下催化加氢生成化合物ⅱ,采用质量比为(1.5~3)的第一催化剂与第二催化剂混合作为化合物i催化加氢的催化剂,第一催化剂与第二催化剂相互配伍,显著提高了化合物ⅱ的收率,从而提高最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率。
附图说明
[0024]
下面结合附图和具体实施方式对本发明作进一步详细的说明。
[0025]
图1为本发明3,4-二丙腈-1,1-二氧四氢噻吩的1h nmr图谱;
[0026]
图2为本发明3,4-二丙腈-1,1-二氧四氢噻吩的
13
c nmr图谱。
具体实施方式
[0027]
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0028]
3,4-二丙腈-1,1-二氧四氢噻吩的合成路线如下:
[0029][0030]
实施例1
[0031]
一种3,4-二丙腈-1,1-二氧四氢噻吩的合成方法,包括以下步骤:
[0032]
a、将300ml乙腈和100g 3,4-二溴噻吩加入反应器中,加入71.82g吡啶,通入氮气,加入0.9g双(三苯基膦)氯化镍、0.1g锌,升温至70℃,滴加48.18g丙烯腈,2h滴毕,保温反应6h,自然冷却至室温,蒸出乙腈,得到72.91g化合物ⅰ,收率为94.86%;
[0033]
b、将50g化合物ⅰ和2.5g催化剂加入干燥的反应釜中,加入450ml甲醇,升温至180℃,通入氢气至压力为2mpa,反应1.5h,反应结束后,自然冷却至室温,泄掉反应釜内氢气,溶液过滤,滤液蒸出甲醇,得到48.3g化合物ⅱ,收率为92.61%;其中,催化剂由质量比为1.5:1的第一催化剂与第二催化剂组成,第一催化剂为钯碳催化剂,钯碳催化剂中钯的质量分数为5%,第二催化剂由质量比为10:7:25的氧化镍、氧化钼及氧化铝载体组成,第二催化剂的比表面积为150~200m2/g,孔容为0.5ml/g;
[0034]
c、将30g化合物ⅱ、90~120ml乙腈加入到反应瓶中,搅拌降温至5~10℃,加入碳酸氢钠水溶液调节ph值至6.8~7.3,降温至0~5℃,滴加90g三氯化钌和0.06~0.18g高碘酸钠混合物,2~3h滴毕,保温30~40min,回至室温,分液,有机相用无水硫酸钠干燥除水后,抽滤,滤液在-0.09mpa、60℃下浓缩,得到31.4g3,4-二丙腈-1,1-二氧四氢噻吩,收率为89.85%。
[0035]
以起始原料3,4-二溴噻吩算,最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率为94.86%
×
92.61%
×
89.85%≈78.93%。
[0036]
实施例2
[0037]
一种3,4-二丙腈-1,1-二氧四氢噻吩的合成方法,包括以下步骤:
[0038]
a、将300ml乙腈和100g 3,4-二溴噻吩加入反应器中,加入81.61g吡啶,通入氮气,
加入0.9g双(三苯基膦)氯化镍、0.1g锌,升温至50℃,滴加54.75g丙烯腈,2.2h滴毕,保温反应7h,自然冷却至室温,蒸出乙腈,得到73.11g化合物ⅰ;收率为95.12%;
[0039]
b、将50g化合物ⅰ和2g催化剂加入干燥的反应釜中,加入500ml甲醇,升温至200℃,通入氢气至压力为2mpa,反应1.1h,反应结束后,自然冷却至室温,泄掉反应釜内氢气,溶液过滤,滤液蒸出甲醇,得到47.6g化合物ⅱ,收率为91.27%;其中,催化剂由质量比为3:1的第一催化剂与第二催化剂组成,第一催化剂为钯碳催化剂,钯碳催化剂中钯的质量分数为5%,第二催化剂由质量比为10:7:25的氧化镍、氧化钼及氧化铝载体组成,第二催化剂的比表面积为150~200m2/g,孔容为0.5ml/g;
[0040]
c、将30g化合物ⅱ、90ml乙腈加入到反应瓶中,搅拌降温至10℃,加入碳酸氢钠水溶液调节ph值至6.8~7.3,降温至5℃,滴加90g三氯化钌和0.06g高碘酸钠混合物,2h滴毕,保温30min,回至室温,分液,有机相用无水硫酸钠干燥除水后,抽滤,滤液在-0.09mpa、60℃下浓缩,得到31.7g3,4-二丙腈-1,1-二氧四氢噻吩,收率为90.71%。
[0041]
以起始原料3,4-二溴噻吩算,最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率为95.12%
×
91.27%
×
90.71%≈78.75%。
[0042]
实施例3
[0043]
一种3,4-二丙腈-1,1-二氧四氢噻吩的合成方法,包括以下步骤:
[0044]
a、将300ml乙腈和100g 3,4-二溴噻吩加入反应器中,加入84.88g吡啶,通入氮气,加入0.9g双(三苯基膦)氯化镍、0.1g锌,升温至60℃,滴加56.94g丙烯腈,2.5h滴毕,保温反应9h,自然冷却至室温,蒸出乙腈,得到73.52g化合物ⅰ;收率为95.65%;
[0045]
b、将50g化合物ⅰ和1.5g催化剂加入干燥的反应釜中,加入600ml甲醇,升温至180℃,通入氢气至压力为2mpa,反应1.3h,反应结束后,自然冷却至室温,泄掉反应釜内氢气,溶液过滤,滤液蒸出甲醇,得到48.5g化合物ⅱ,收率为93%;其中,催化剂由质量比为2:1的第一催化剂与第二催化剂组成,第一催化剂为钯碳催化剂,钯碳催化剂中钯的质量分数为5%,第二催化剂由质量比为10:7:25的氧化镍、氧化钼及氧化铝载体组成,第二催化剂的比表面积为150~200m2/g,孔容为0.5ml/g;
[0046]
c、将30g化合物ⅱ、100ml乙腈加入到反应瓶中,搅拌降温至5℃,加入碳酸氢钠水溶液调节ph值至6.8,降温至0℃,滴加90g三氯化钌和0.15g高碘酸钠混合物,2.5h滴毕,保温35min,回至室温,分液,有机相用无水硫酸钠干燥除水后,抽滤,滤液在-0.09mpa、60℃下浓缩,得到31.2g3,4-二丙腈-1,1-二氧四氢噻吩,收率为89.27%。
[0047]
以起始原料3,4-二溴噻吩算,最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率为95.65%
×
93%
×
89.27%≈79.41%。
[0048]
实施例4
[0049]
一种3,4-二丙腈-1,1-二氧四氢噻吩的合成方法,包括以下步骤:
[0050]
a、将300ml乙腈和100g 3,4-二溴噻吩加入反应器中,加入84.88g吡啶,通入氮气,加入1.8g双(三苯基膦)氯化镍、0.2g锌,升温至60℃,滴加56.94g丙烯腈,2.5h滴毕,保温反应9h,自然冷却至室温,蒸出乙腈,得到72.87g化合物ⅰ;收率为94.81%;
[0051]
b、将50g化合物ⅰ和1g催化剂加入干燥的反应釜中,加入500ml甲醇,升温至180℃,通入氢气至压力为2mpa,反应1.5h,反应结束后,自然冷却至室温,泄掉反应釜内氢气,溶液过滤,滤液蒸出甲醇,得到48.2g化合物ⅱ,收率为92.42%;其中,催化剂由质量比为3:1的
第一催化剂与第二催化剂组成,第一催化剂为钯碳催化剂,钯碳催化剂中钯的质量分数为5%,第二催化剂由质量比为10:7:25的氧化镍、氧化钼及氧化铝载体组成,第二催化剂的比表面积为150~200m2/g,孔容为0.5ml/g;
[0052]
c、将30g化合物ⅱ、120ml乙腈加入到反应瓶中,搅拌降温至10℃,加入碳酸氢钠水溶液调节ph值至7,降温至0℃,滴加90g三氯化钌和0.06g高碘酸钠混合物,2h滴毕,保温30min,回至室温,分液,有机相用无水硫酸钠干燥除水后,抽滤,滤液在-0.09mpa、60℃下浓缩,得到31.8g3,4-二丙腈-1,1-二氧四氢噻吩,收率为90.99%。
[0053]
以起始原料3,4-二溴噻吩算,最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率为94.81%
×
92.42%
×
90.99%≈79.73%。
[0054]
实施例5
[0055]
一种3,4-二丙腈-1,1-二氧四氢噻吩的合成方法,包括以下步骤:
[0056]
a、将300ml乙腈和100g 3,4-二溴噻吩加入反应器中,加入84.88g吡啶,通入氮气,加入1g双(三苯基膦)氯化镍、0.1g锌,升温至60℃,滴加56.94g丙烯腈,2.5h滴毕,保温反应9h,自然冷却至室温,蒸出乙腈,得到73.08g化合物ⅰ;收率为95.08%;
[0057]
b、将50g化合物ⅰ和2.5g催化剂加入干燥的反应釜中,加入500ml甲醇,升温至200℃,通入氢气至压力为2mpa,反应1h,反应结束后,自然冷却至室温,泄掉反应釜内氢气,溶液过滤,滤液蒸出甲醇,得到47.8g化合物ⅱ,收率为91.66%;其中,催化剂由质量比为1.5:1的第一催化剂与第二催化剂组成,第一催化剂为钯碳催化剂,钯碳催化剂中钯的质量分数为5%,第二催化剂由质量比为10:7:25的氧化镍、氧化钼及氧化铝载体组成,第二催化剂的比表面积为150~200m2/g,孔容为0.5ml/g;
[0058]
c、将30g化合物ⅱ、100ml乙腈加入到反应瓶中,搅拌降温至10℃,加入碳酸氢钠水溶液调节ph值至7.3,降温至5℃,滴加90g三氯化钌和0.18g高碘酸钠混合物,2.5h滴毕,保温40min,回至室温,分液,有机相用无水硫酸钠干燥除水后,抽滤,滤液在-0.09mpa、60℃下浓缩,得到32.1g3,4-二丙腈-1,1-二氧四氢噻吩,收率为91.85%。
[0059]
以起始原料3,4-二溴噻吩算,最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率为95.08%
×
91.66%
×
91.85%≈80.05%。
[0060]
对比例1
[0061]
本对比例与实施例1的区别在于步骤a中加入0.88g双(三苯基膦)氯化镍、0.12g锌;其余步骤同实施例1;结果步骤a中得到69.58g化合物ⅰ,收率为90.53%;以起始原料3,4-二溴噻吩算,最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率为65.53%。
[0062]
对比例2
[0063]
本对比例与实施例1的区别在于步骤a中加入0.83g双(三苯基膦)氯化镍、0.17g锌;其余步骤同实施例1;结果步骤a中得到67.31g化合物ⅰ,收率为87.58%;以起始原料3,4-二溴噻吩算,最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率为72.88%。
[0064]
对比例3
[0065]
本对比例与实施例1的区别在于步骤b中催化剂为钯碳催化剂,加入量为2.5g,钯碳催化剂中钯的质量分数为5%,其余步骤同实施例1,结果步骤b中得到43.7g化合物ⅱ,收率为83.80%;以起始原料3,4-二溴噻吩算,最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率为67.12%。
[0066]
对比例4
[0067]
本对比例与实施例1的区别在于步骤b中催化剂为第二催化剂,加入量为2.5g,由质量比为10:7:25的氧化镍、氧化钼及氧化铝载体组成,第二催化剂的比表面积为150~200m2/g,孔容为0.5ml/g,其余步骤同实施例1,结果步骤b中得到40.2g化合物ⅱ,收率为77.08%;以起始原料3,4-二溴噻吩算,最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率为65.70%。
[0068]
与实施例1相比,对比例1~2的步骤a中化合物ⅰ的收率大大降低,说明步骤a中采用质量比为(8-10):1的双(三苯基膦)氯化镍与锌混合作为催化剂,特定质量比的双(三苯基膦)氯化镍与锌协同作用,显著提高了化合物i的收率,从而提高最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率。
[0069]
与实施例1相比,对比例3~4的步骤b中化合物ⅱ的收率大大降低,说明步骤b中采用质量比为(1.5~3)的第一催化剂与第二催化剂混合作为化合物i催化加氢的催化剂,第一催化剂与第二催化剂相互配伍,显著提高了化合物ⅱ的收率,从而提高最终产物3,4-二丙腈-1,1-二氧四氢噻吩的总收率。
[0070]
最终产物3,4-二丙腈-1,1-二氧四氢噻吩的1h nmr图谱、
13
c nmr图谱如图1~2所示。
[0071]
以上仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。