脒/胍基修饰的真菌cyp51抑制剂衍生物及其制备方法和应用
技术领域
1.本发明属于医药技术领域,涉及脒/胍基修饰的真菌cyp51抑制剂衍生物及其制备方法和应用。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.临床上应用于抵抗真菌感染的药物主要有唑类、多烯类、棘白菌素类、烯丙胺类及嘧啶类,其中唑类药物如氟康唑、伊曲康唑、伏立康唑等在临床上针对于系统性真菌感染疾病的应用最为普遍。而由于抗生素误用或滥用,导致真菌的耐药性增加,限制了目前临床应用的抗生素的治疗效果,给系统性真菌感染治疗造成了巨大挑战。因此需要寻找新抗真菌药物以解决抗真菌药物耐药性的问题。
技术实现要素:
4.为了解决现有技术的不足,本发明的目的是提供脒/胍基修饰的真菌cyp51抑制剂衍生物及其制备方法和应用。
5.为了实现上述目的,本发明的技术方案为:
6.一方面,一种脒/胍基修饰的真菌cyp51抑制剂衍生物,其化学结构如式i所示,
[0007][0008]
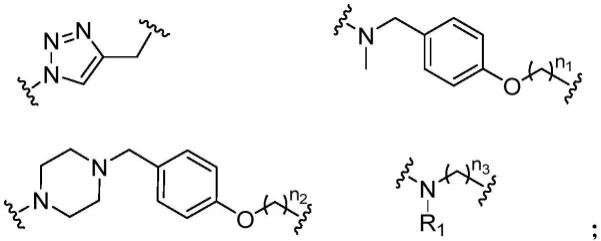
其中,r、x均为连接基团,r选自以下结构中的一种:
[0009][0010]
n1为0~10的自然数,n2为0~10的自然数,n3为0~10的自然数,
[0011]
r1选自氢、取代或未取代烷基、取代或未取代环烷基;
[0012]
x选自不存在、取代或未取代苯基;
[0013]
a为0或1。
[0014]
当a为0时,x直接与脒基连接,即修饰基团为脒基,其通式为:
[0015][0016]
当a为1时,修饰基团为胍基,其通式为:
[0017][0018]
当x不存在时,连接基团r直接与脒基或胍基连接,即通式为:
[0019][0020]
r1中取代烷基中的取代基团为卤素,例如f、cl、br、i。
[0021]
r1中取代环烷基中的取代基团为卤素,例如f、cl、br、i。
[0022]
当n1、n2、n3分别为0时,r的结构式分别为:
[0023][0024]
x中取代苯基中的取代基团为羟基、氨基、巯基及卤素(例如f、cl、br、i)。取代苯基中的两个连接位点可以均在苯环上;也可以一个连接位点在苯环上,另一个连接位点在取代基上,例如当取代基为羟基的时,x可以分别为等。
[0025]
在一些实施例中,r1选自氢、取代或未取代c1~20烷基、取代或未取代c3~20环烷基。
[0026]
在一些实施例中,r1选自h、甲基、乙基、正丙基、正丁基、异丙基、环丙烷基、异丁基。
[0027]
在一些实施例中,n1为1~10的自然数,n2为1~10的自然数,n3为1~10的自然数。
[0028]
在一些实施例中,其通式为:
[0029][0030]
在一些实施例中,衍生物为化合物a,化合物a的结构如下:
[0031][0032]
在一些实施例中,衍生物为化合物b,化合物b的结构如下:
[0033][0034]
其中,n3’
为1~8的自然数;r1选自h、甲基、乙基、正丙基、正丁基、异丙基、环丙烷基、异丁基;x’为连接基团,x’为
[0035]
x’为时,苯环的连接位点直接连接脒基。
[0036]
在一些实施例中,衍生物为化合物c,化合物c的结构如下:
[0037][0038]
其中,n’为1~8的自然数;x’为连接基团,x’为y为连接基团,y为
[0039]
x’为时,苯环的连接位点直接连接脒基。
[0040]
在一些实施例中,包括以下任一一种结构的化合物:
[0041][0042]
另一方面,一种脒/胍基修饰的真菌cyp51抑制剂衍生物的制备方法,包括按照如下反应路线制备化合物a1的步骤:
[0043][0044]
第一步,将化合物1溶于干燥的dmf中,加入试剂(boc)2o、dmap、dipea,反应得到化合物2;
[0045]
第二步,将化合物2溶于甲醇中,加入试剂naoh、水,反应得到化合物3;
[0046]
第三步,将化合物3溶于干燥的乙腈中,加入试剂炔丙基溴、k2co3,反应得到化合物4;
[0047]
第四步,将化合物4溶于异丙醇中,加入试剂叠氮中间体m1、五水硫酸铜、抗坏血酸钠,反应得到化合物5;
[0048]
第五步,将化合物5溶于三氟乙酸/二氯甲烷中,反应得到化合物a1。
[0049]
包括按照如下反应路线制备化合物b1-b3的步骤:
[0050][0051]
第一步,将化合物3溶于干燥的乙腈中,加入试剂br(ch2)
n br、k2co3,反应得到化合物6;试剂br(ch2)
n br中n为3或5或8;
[0052]
第二步,将化合物6溶于干燥的dmf中,加入试剂叠氮化钠,反应得到化合物7;
[0053]
第三步,将化合物7溶于干燥的thf中,加入试剂三苯基膦、水,反应得到化合物8;
[0054]
第四步,将化合物8溶于干燥的乙醇中,加入试剂环氧中间体m2、三乙胺,反应得到化合物9;
[0055]
第五步,将化合物9溶于三氟乙酸/二氯甲烷中,反应得到化合物b1-b3。
[0056]
包括按照如下反应路线制备化合物b4-b5的步骤:
[0057][0058]
第一步,将环氧中间体m2溶于干燥的乙醇中,加入试剂三乙胺、ch3nh(ch2)nnhboc,反应得到化合物10;ch3nh(ch2)nnhboc中n为3或5;
[0059]
第二步,将化合物10溶于三氟乙酸/二氯甲烷中,反应得到化合物11;
[0060]
第三步,将化合物11溶于干燥的thf中,加入试剂n,n'-二-boc-1h-1-胍基吡唑、三乙胺,反应得到化合物12;
[0061]
第四步,将化合物12溶于三氟乙酸/二氯甲烷中,反应得到化合物b4-b5。
[0062]
包括按照如下反应路线制备化合物c1-c4的步骤:
[0063][0064]
第一步,将化合物13溶于干燥的乙腈中,加入试剂br(ch2)nnhboc、k2co3,反应得到化合物14;试剂br(ch2)nnhboc中n为3或6;
[0065]
第二步,将化合物14溶于干燥的甲醇中,加入试剂盐酸甲胺或哌嗪、钛酸四异丙酯、三乙胺,室温搅拌反应,加入试剂硼氢化钠,继续室温搅拌反应得到化合物15;
[0066]
第三步,将化合物15溶于干燥的乙醇中,加入试剂环氧中间体m2、三乙胺,反应得到化合物16;
[0067]
第四步,将化合物16溶于三氟乙酸/二氯甲烷中,反应得到化合物17;
[0068]
第五步,将化合物17溶于干燥的thf中,加入试剂n,n'-二-boc-1h-1-胍基吡唑、三乙胺,反应得到化合物18;
[0069]
第六步,将化合物18溶于三氟乙酸/二氯甲烷中,反应得到化合物c1-c4。
[0070]
包括按照如下反应路线制备化合物c5-c6的步骤:
[0071][0072]
第一步,将化合物19溶于干燥的乙腈中,加入试剂对羟基苯甲醛、k2co3,反应得到化合物20;
[0073]
第二步,将化合物20溶于干燥的甲醇中,加入试剂盐酸甲胺或哌嗪、钛酸四异丙酯、三乙胺,室温搅拌反应,加入试剂硼氢化钠,继续室温搅拌反应得到化合物21;
[0074]
第三步,将化合物21溶于干燥的乙醇中,加入试剂环氧中间体m2、三乙胺,反应得到化合物22;
[0075]
第四步,将化合物22溶于三氟乙酸/二氯甲烷的混合溶剂中,反应得到化合物c5-c6。
[0076]
第三方面,一种药物组合物,其含有上述脒/胍基修饰的真菌cyp51抑制剂衍生物或其药学上可接受的盐。
[0077]
本发明所述的药学上可接受的盐可以为盐酸盐、硫酸盐、枸橼酸盐等。
[0078]
第四方面,一种药物制剂,包括活性成分和药学上可接受的辅料,所述活性成分为上述脒/胍基修饰的真菌cyp51抑制剂衍生物或上述药物组合物。
[0079]
本发明所述的药学上可接受的辅料可以为粘合剂(如阿拉伯胶、聚乙烯吡咯烷酮、糖浆等),可以为填充剂(如磷酸钙、玉米淀粉等),可以为崩解剂(如马铃薯淀粉等),可以为润滑剂(如硬脂酸镁、聚乙二醇等)。
[0080]
本发明所述药物制剂的给药剂型可以为片剂、丸剂、胶囊、溶液剂等。
[0081]
第五方面,一种上述脒/胍基修饰的真菌cyp51抑制剂衍生物、药物组合物或药物制剂在制备抗真菌类药物中的应用。
[0082]
本发明所述的真菌为临床上常见的致病真菌,包括:白色念珠菌、热带念珠菌、光滑念珠菌、克柔念珠菌、近平滑念珠菌、耳念珠菌等。
[0083]
由于采用以上技术方案,本发明具有以下优点:
[0084]
本发明的脒/胍基修饰的真菌cyp51抑制剂抗衍生物通过在传统cyp51抑制剂的关键药效团结构中引入苯脒/胍基,获得了一系列脒/胍基修饰的cyp51抑制剂衍生物,这些化合物不仅对唑类敏感的菌株有效,同样对唑类耐药的菌株也具有十分显著的抗真菌活性。
具体实施方式
[0085]
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
[0086]
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包
括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
[0087]
为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例与对比例详细说明本发明的技术方案。
[0088]
实施例
[0089]
化合物a的制备:
[0090][0091]
试剂与条件:(a)(boc)2o,dmap,dipea,dmf,室温,12h;(b)naoh,h2o,meoh,室温,2h;(c)炔丙基溴,k2co3,mecn,80℃,10h;(d)l-抗坏血酸钠盐,cuso4,t-buoh,室温,6h;(e)tfa,ch2cl2,室温,2h。
[0092]
化合物a1的制备方法:
[0093]
第一步,将500mg化合物1溶于20ml干燥的dmf中,加入1.26g试剂(boc)2o、35mg dmap、1.9ml dipea,室温反应12h,加水析出固体,过滤得到化合物2。
[0094]
第二步,将800mg化合物2溶于15ml甲醇中,加入15ml 10%的naoh溶液,室温反应2h,旋蒸除掉大部分甲醇,加水,以乙酸乙酯萃取多次,合并有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,得到化合物3。
[0095]
第三步,将200mg化合物3溶于15ml干燥的乙腈中,加入0.3ml试剂炔丙基溴、460mg k2co3,80℃反应10h,加水淬灭反应,以乙酸乙酯萃取两次,合并有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以石油醚/乙酸乙酯(3/1,v/v)为流动相经硅胶柱层析纯化得到化合物4。
[0096]
第四步,将200mg化合物4溶于15ml异丙醇/水(4:1)的混合溶剂中,加入205mg试剂叠氮中间体m1、10mg cuso4、55mg抗坏血酸钠,室温反应6h,加水淬灭反应,以二氯甲烷萃取两次,合并有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以二氯甲烷/甲醇(20/1,v/v)为流动相经硅胶柱层析纯化得到化合物5。
[0097]
第五步,将300mg化合物5溶于6ml三氟乙酸/二氯甲烷(2/1,v/v)的混合溶剂中,室温反应2h,减压除去有机溶剂,残余物以饱和的碳酸氢钠溶液中和,以乙酸乙酯萃取混合物,无水naso4干燥,过滤,滤液减压除去有机溶剂,得到化合物a1。
[0098]
化合物b1-b3的制备:
[0099][0100]
试剂与条件:(a)br(ch2)nbr,k2co3,mecn 80℃,10h;(b)nan3,dmf,50℃,12h;(c)tpp,h2o,thf,室温,4h;(d)et3n,etoh,70℃,10h;(e)tfa,ch2cl2,室温,2h.
[0101]
化合物b1的制备方法:
[0102]
第一步,将200mg化合物3溶于8ml干燥的乙腈中,加入250μl试剂br(ch2)3br、110mg k2co3,80℃反应10h,加水淬灭反应,以乙酸乙酯萃取两次,合并有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以石油醚/乙酸乙酯(3/1,v/v)为流动相经硅胶柱层析纯化得到化合物6。
[0103]
第二步,将280mg化合物6溶于8ml干燥的dmf中,加入152mg试剂叠氮化钠,50℃反应12h,加入乙酸乙酯稀释,以饱和食盐水洗涤三次,取有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以石油醚/乙酸乙酯(3/1,v/v)为流动相经硅胶柱层析纯化得到化合物7。
[0104]
第三步,将200mg化合物7溶于6ml干燥的thf中,加入180mg试剂三苯基膦、120μl水,室温反应4h,旋蒸除去溶剂,残余物以二氯甲烷/甲醇(10/1,v/v)为流动相经硅胶柱层析纯化得到化合物8。
[0105]
第四步,将160mg化合物8溶于5ml干燥的乙醇中,加入140mg试剂环氧中间体、240μl三乙胺,70℃反应10h,加入乙酸乙酯稀释反应液,以饱和食盐水洗涤,取有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以二氯甲烷/甲醇(15/1,v/v)为流动相经硅胶柱层析纯化得到化合物9。
[0106]
第五步,将200mg化合物9溶于4ml三氟乙酸/二氯甲烷(2/1,v/v)的混合溶剂中,室温反应2h,减压除去有机溶剂,残余物以饱和的碳酸氢钠溶液中和,以乙酸乙酯萃取混合物,无水naso4干燥,过滤,滤液减压除去有机溶剂,得到化合物b1。
[0107]
化合物b2与b3的制备方法:制备方法与化合物b1相同,不同之处在于第一步加入试剂br(ch2)3br分别替换为br(ch2)5br,br(ch2)8br,经五步反应最终得到化合物b2(n为5),b3(n为8)。
[0108]
化合物b4-b5的制备:
[0109][0110]
试剂与条件:(a)ch3nh(ch2)nnhboc,et3n,etoh,70℃,10h;(b)tfa,ch2cl2,室温,2h;(c)boc-吡唑,et3n,室温,12h;(d)tfa,ch2cl2,室温,2h.
[0111]
化合物b4的制备方法:
[0112]
第一步,将300mg环氧中间体m2溶于10ml干燥的乙醇中,加入238mg试剂ch3nh(ch2)3nhboc、520μl三乙胺,70℃反应10h,加入乙酸乙酯稀释反应液,以饱和食盐水洗涤,取有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以二氯甲烷/甲醇(40/1,v/v)为流动相经硅胶柱层析纯化得到化合物10。
[0113]
第二步,将400mg化合物10溶于10ml三氟乙酸/二氯甲烷(2/1,v/v)的混合溶剂中,室温反应2h,减压除去有机溶剂,残余物以饱和的碳酸氢钠溶液中和,以乙酸乙酯萃取混合物,无水naso4干燥,过滤,滤液减压除去有机溶剂,得到化合物11。
[0114]
第三步,将200mg化合物11溶于6ml干燥的thf中,加入试剂228mg n,n'-二-boc-1h-1-胍基吡唑、400μl三乙胺,室温反应12h,加入乙酸乙酯稀释反应液,以饱和食盐水洗涤,取有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以石油醚/乙酸乙酯(3/1,v/v)为流动相经硅胶柱层析纯化到化合物12。
[0115]
第四步,将300mg化合物12溶于5ml三氟乙酸/二氯甲烷(2/1,v/v)的混合溶剂中,室温反应2h,减压除去有机溶剂,残余物以饱和的碳酸氢钠溶液中和,以乙酸乙酯萃取混合物,无水naso4干燥,过滤,滤液减压除去有机溶剂,得到化合物b4。
[0116]
化合物b5的制备方法:制备方法与b4相同,不同之处在于第一步加入试剂ch3nh(ch2)3nhboc替换为ch3nh(ch2)5nhboc,经后续三步反应最终得到化合物b5(n为5)。
[0117]
化合物c1-c4的制备:
[0118][0119]
试剂与条件:(a)br(ch2)nnhboc,k2co3,mecn,80℃,10h;(b)钛酸异丙酯,et3n,meoh,室温,12h;nabh4,室温,0.5h;(c)et3n,etoh,70℃,10h;(d)tfa,ch2cl2,室温,2h;(e)
boc-吡唑,et3n,室温,12h;(f)tfa,ch2cl2,室温,2h.
[0120]
化合物c1的制备方法:
[0121]
第一步,将200mg化合物13溶于10ml干燥的乙腈中,加入585mg试剂br(ch2)3nhboc、678mg k2co3,80℃反应10h,加水淬灭反应,以乙酸乙酯萃取两次,合并有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以石油醚/乙酸乙酯(3/1,v/v)为流动相经硅胶柱层析纯化得到化合物14。
[0122]
第二步,将354mg化合物14溶于10ml干燥的甲醇中,加入128mg试剂盐酸甲胺、720mg钛酸四异丙酯、528μl三乙胺,室温搅拌反应12h;冰浴加入422mg试剂硼氢化钠,继续室温搅拌反应0.5h。向反应液加入水,抽滤,乙酸乙酯清洗滤饼,收集滤液,以乙酸乙酯萃取,取有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物即为化合物15。
[0123]
第三步,将200mg化合物15溶于8ml干燥的乙醇中,加入200mg试剂环氧中间体m2、280μl三乙胺,70℃反应10h,加入乙酸乙酯稀释反应液,以饱和食盐水洗涤,取有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以二氯甲烷/甲醇(20/1,v/v)为流动相经硅胶柱层析纯化得到化合物16。
[0124]
第四步,将310mg化合物16溶于6ml三氟乙酸/二氯甲烷(2/1,v/v)的混合溶剂中,室温反应2h,减压除去有机溶剂,残余物以饱和的碳酸氢钠溶液中和,以乙酸乙酯萃取混合物,无水naso4干燥,过滤,滤液减压除去有机溶剂,得到化合物17。
[0125]
第五步,将250mg化合物17溶于5ml干燥的thf中,加入150mg试剂n,n'-二-boc-1h-1-胍基吡唑、400μl三乙胺,室温反应12h,减压除去有机溶剂,残余物中加入乙酸乙酯,以饱和食盐水洗涤,取有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以二氯甲烷/甲醇(20/1,v/v)为流动相经硅胶柱层析纯化得到化合物得到化合物18。
[0126]
第六步,将50mg化合物18溶于1ml三氟乙酸/二氯甲烷(2/1,v/v)的混合溶剂中,室温反应2h,减压除去有机溶剂,残余物以饱和的碳酸氢钠溶液中和,以乙酸乙酯萃取混合物,无水naso4干燥,过滤,滤液减压除去有机溶剂,得到化合物c1。
[0127]
化合物c2的制备:c2的制备方法与化合物c1相同,不同之处在于第二步加入试剂盐酸甲胺替换为哌嗪,再经后续反应最终得到化合物c2(n为3)。
[0128]
化合物c3的制备:c3的制备方法与化合物c1相同,不同之处在于第一步加入试剂br(ch2)3nhboc替换为br(ch2)6nhboc,再经后续反应最终得到化合物c3(n为6)。
[0129]
化合物c4的制备:c4的制备方法与化合物c1相同,不同之处在于第一步加入试剂br(ch2)3nhboc替换为br(ch2)6nhboc,第二步加入试剂盐酸甲胺替换为哌嗪,再经后续反应最终得到化合物c6(n为6)。
[0130]
化合物c5-c6的制备:
[0131][0132]
试剂与条件:(a)k2co3,mecn,80℃,10h;(b)钛酸异丙酯,et3n,meoh,室温,12h;
nabh4,室温,0.5h;(c)et3n,etoh,70℃,10h;(d)tfa,ch2cl2,室温,2h.
[0133]
化合物c5的制备方法:
[0134]
第一步,将200mg化合物19溶于5ml干燥的乙腈中,加入75mg试剂对羟基苯甲醛、300mg k2co3,80℃反应10h,加水淬灭反应,以乙酸乙酯萃取两次,合并有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以石油醚/乙酸乙酯(2/1,v/v)为流动相经硅胶柱层析纯化得到化合物20。
[0135]
第二步,将150mg化合物20溶于5ml干燥的甲醇中,加入38mg试剂盐酸甲胺、214mg钛酸四异丙酯、157μl三乙胺,室温搅拌反应12h,加入98mg试剂硼氢化钠,继续室温搅拌反应0.5h。向反应液加入水,抽滤,乙酸乙酯清洗滤饼,收集滤液,以乙酸乙酯萃取,取有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物即为化合物21。
[0136]
第三步,将52mg化合物21溶于1ml干燥的乙醇中,加入36mg试剂环氧中间体m2、50μl三乙胺,70℃反应10h,加入乙酸乙酯稀释反应液,以饱和食盐水洗涤,取有机相,无水naso4干燥,过滤,滤液减压除去有机溶剂,残余物以二氯甲烷/甲醇(20/1,v/v)为流动相经硅胶柱层析纯化得到化合物22。
[0137]
第四步,将60mg化合物22溶于1ml三氟乙酸/二氯甲烷(2/1,v/v)的混合溶剂中,室温反应2h,减压除去有机溶剂,残余物以饱和的碳酸氢钠溶液中和,以乙酸乙酯萃取混合物,无水naso4干燥,过滤,滤液减压除去有机溶剂,得到化合物c5。
[0138]
化合物c6的制备:c6的制备方法与化合物c5相同,不同之处在于第二步加入试剂盐酸甲胺替换为哌嗪,再经后续反应最终得到化合物c4(n为6)。
[0139]
上述制备的本发明部分优选化合物的化学结构式、nmr数据详见表1。
[0140]
表1本发明化合物的nmr数据
[0141]
[0142][0143][0144]
效果实施例
[0145]
真菌生长抑制试验
[0146]
使用上述制备的化合物a1、b1~b5、c1~c6进行真菌生长抑制试验,菌株选用白色念珠菌包括:野生型sc5314、基因工程化改造的外排泵过表达耐药菌株(yem13、yem15)、临床分离的外排泵高表达的耐药白色念珠菌(g5、dsy296),其他念珠菌包括:耳念珠菌0029、热带念珠菌ct-q-2、光滑念珠菌cg1、克柔念珠菌ck9、近平滑念珠菌cp001,抗真菌药物氟康唑(flc)作为阳性对照药。
[0147]
材料及仪器:移液枪(thermo fisher scientific),96孔板(nest biotechnology),涡旋振荡器(scientific industries vortex-2 genie),ep管(南通顺丰医疗器械有限公司sf-ep-002),双人单面超净工作台(苏州净化设备有限公司sw-cj-jc型),恒温摇床(eppendorf nbs innova 42型),离心机(eppendorf minispin plus),高压蒸汽灭菌锅(sanyo mls-3750),流式细胞仪(bd facscalibur)。
[0148]
培养基配置:
[0149]
ypd液体培养基:蛋白胨20g,酵母淀粉10g,加三蒸水800ml溶解,取葡萄糖20g,加三蒸水200ml。将上述两种溶液进行高温高压灭菌(121℃,20min)后混合即得。
[0150]
rpmi-1640液体培养基:rpmi-1640固体粉末(gibco brl)10g,3-吗啉基丙磺酸34.5g,加三蒸水900ml充分溶解,1m naoh调ph至7.0(25℃),三蒸水定容至1000ml,0.22μm微孔滤膜过滤除菌,分装后于4℃保存备用。
[0151]
pbs缓冲液的配置:称取8g nacl、0.2g kcl、1.44g na2hpo4和0.24g kh2po4,溶于800ml蒸馏水中,用hcl调节溶液的ph值至7.4,最后加蒸馏水定容至1l,高压灭菌,置于4℃保存备用。
[0152]
待测药物的配制:待测药物用dmso溶解,配制成10mg/ml的溶液,置于-20℃冰箱储存备用。
[0153]
菌悬液的制备:用接种圈从4℃保存的ypd培养基上挑取单克隆菌株,接种至1ml ypd培养液中,于30℃,200rpm振荡培养,培养大约16h,使真菌处于指数生长期后期。取该菌液至1.5ml ep管,离心(13400rpm,1min),弃去上清,用1ml pbs缓冲液将菌体吹打混悬,用pbs缓冲溶液吹洗2次,用1ml rpmi 1640培养液吹打混悬,采用比浊法将菌浓度调整至约1
×
103cfu/ml。
[0154]
加药及检测:将配制好的菌悬液加入96孔板中,第1列加入200μl,2至12列加入l00μl。每个待测药物(10mg/ml)取2.56μl于第一列菌悬液中,倍半稀释11个浓度梯度,即终浓度为128μg/ml-0.125μg/ml,第12列不加药,作为对照。放入35℃恒温培养箱中孵育24h,用酶标仪测定od
600
。以阴性对照孔的od值设置为100%,最低抑菌浓度(mic
80
)为抑制真菌80%以上生长(od值降低80%以上)的最低化合物浓度。对于抗真菌活性低于0.125μg/ml的化合物,进一步设置药物浓度0.125μg/ml-0.0039μg/ml,获得准确的mic
80
值,实验结果如表2所示。测试结果表明部分氟康唑衍生物如c3和c5优于氟康唑的抗真菌活性,并且对外排泵高表达耐药菌株以及超级耐药真菌耳念珠菌具有非常强的抗真菌活性。
[0155]
表2化合物体外抗真菌活性测试
[0156][0157]
注:
“‑”
表示未对该菌株进行活性测试,flc为氟康唑。
[0158]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。