一种kras突变体g12c抑制剂及其制备方法和应用
技术领域
1.本发明涉及药物化学领域,具体涉及一种kras突变体g12c抑制剂及其制备方法和应用。
背景技术:
2.ras基因是首个被发现的人类肿瘤基因,其编码的ras蛋白为一类分布于质膜胞质侧的球状单体gtp结合蛋白(mw=21kda),结合gtp时为活化状态,而结合gdp时为失活状态。ras蛋白在鸟嘌呤核苷酸交换因子(gef)的催化下结合gtp而激活,而gtp酶激活蛋白(gaps)催化与ras结合的gtp水解成为gdp终止活性状态以抑制ras活性。ras蛋白通过其在活性(gtp结合型)和非活性(gdp结合型)状态间循环,将从多种酪氨酸激酶接收的上游信号转导至下游效应物以调节细胞增殖、存活、迁移和凋亡等过程。由于ras蛋白在许多重要细胞信号网络的轴上处于中心位置,且这些信号与多种肿瘤标志物相关联,因此过度活化的ras信号转导可能最终导致肿瘤发生。
3.在ras家族成员中,致癌突变最常见于kras(85%),而nras(12%)和hras(3%)则较为少见,且kras突变常见于胰腺癌(95%)、结直肠癌(45%)、肺癌(25%)、胆囊癌及甲状腺癌等,kras的异常表达占所有癌症的比例高达20%,而80%的kras突变为第12位密码子单个氨基酸替换的错义突变,如kras g12c、g12v和g12d。其中kras g12c突变占所有kras突变的12%,但其在肺癌种所占比例较高,尤其是非小细胞肺癌(14%),基因组学研究表明,肺癌kras突变(包括g12c)与其它已知的致癌基因突变相斥,如egfr、alk及braf,这表明kras突变在肺癌中的独特性且其可作为肿瘤预后的一个重要指标。
4.由于kras其无明显结合位点的蛋白结构,经过四十多年的研究,虽然人们对ras通路致病机制有了更为深入的了解,但临床上仍没有kras抑制剂呈现出足够的安全性和有效性。近年来,针对kras突变体的共价抑制剂研究的突破让通过异构位点(allosteric)靶向kras突变体成为可能。在kras g12c突变体中,与错义突变替换的半胱氨酸共价结合的小分子更倾向与结合gdp的kras蛋白相结合以降低gtp与kras的亲和力,同时能够阻碍gef催化gtp替换gdp,将kras g12c突变体锁死在失活状态。利用这种策略,目前已经有美国amgen公司的amg510和美国mirati公司的mrtx-849进入了临床研究。除此之外,各药物研发公司也相继研发并发表了多篇专利,比如wo2019150305、wo2020106640、wo2020146613、wo2019215203、wo2020081282、wo2020085493、cn110256421a、cn113045565a、cn112312901a等所述的kras g12c抑制剂。
5.因此继续开发新颖的kras g12c抑制剂,且能够表现出治疗kras g12c介导肿瘤的有效性、稳定性及安全性,在kras突变引起的肿瘤的治疗领域是迫切需要和极具意义的。
技术实现要素:
6.针对上述不足,本发明提供了一种kras突变体g12c抑制剂及其制备方法和应用。所述的kras突变体g12c抑制剂为萘啶甲腈类衍生物、其药学上可接受的盐、其互变异构体
或其立体异构体,其具有较好的抗肿瘤有效性、稳定性及安全性,具有广泛的抗肿瘤药物应用前景。
7.为了实现上述发明目的,本发明的技术方案如下:
8.一方面,本发明提供了一种kras突变体g12c抑制剂,所述的抑制剂为萘啶甲腈类衍生物、其药学上可接受的盐、其互变异构体或其立体异构体。
9.具体地,所述的萘啶甲腈类衍生物的结构如式(i)所示。
[0010][0011]
其中,
[0012]
l1为o、s或crarb。
[0013]
ra和rb分别独立选自h、卤素、羟基、c1-c3烷基、c3-7的环烷基或3-7元的杂环烷基,而且ra和rb连同它们所附接的原子可以进一步形成c3-7的环烷基或c3-7的杂环烷基。
[0014]
r1为6-12元的芳基或5-12元的杂芳基,所述的芳基或杂芳基可以任选进一步被1-5个rc所取代;
[0015]
rc选自h、卤素、羟基、nrarb、c1-c3烷基、氰基、烷氧基、=o、c3-7环烷基、3-8元杂环基、c2-c3烯基、c2-c3炔基、c(o)r
c1
、c(o)nrarb或nrac(o)rb,其中所述的烷基、环烷基、杂环烷基、烯基、ra或rb可以任选进一步被1-3个r
c2
所取代;
[0016]rc1
选自c1-c5烷基、c3-7的环烷基或3-7元的杂环基;
[0017]rc2
选自h、卤素、羟基、氨基、c1-c3烷基、烷氧基或氰基。
[0018]
l2为o、s或crarb。
[0019]
r2为c1-c3烷基、c3-7的环烷基、3-8元的杂环基、6-12元的芳基或5-12元的杂芳基,其中所述的烷基、环烷基、杂环烷基、芳基或杂芳基可以任选进一步被1-5个rc所取代。
[0020]
l3选自c3-8的环烷基、c3-8螺环烷基、c3-8稠环烷基、c3-8桥环烷基、3-8元的杂环烷基、3-8元的螺杂环烷基、3-8元的稠杂环烷基、6-12元的芳基或5-12元的杂芳基,其中所述的环烷基、稠环烷基、桥环烷基、杂环基、螺杂环烷基、稠杂环烷基、芳基、杂芳基可以任选进一步被1-4个rc所取代。
[0021]
r3选自c(o)rd;
[0022]
rd选自c2-4烯基或c2-4炔基,其中所述的烯基或炔基可以任选进一步被1-3个r
d1
所取代;
[0023]rd1
选自h、卤素、羟基、nrarb、c1-c3烷基、c3-7环烷基、3-7元杂环基或5-12元的杂芳基。
[0024]
r4选自h、卤素、c1-c3烷基、氰基或c1-2的烷氧基。
[0025]
进一步具体地,所述的萘啶甲腈类衍生物的结构如式(ii)所示。
[0026][0027]
其中,
[0028]
n=0-4(例如n=0、n=1、n=2、n=3、n=4)。
[0029]
r1为6-12元的芳基或5-12元的杂芳基,所述的芳基或杂芳基可以任选进一步被1-5个rc所取代;
[0030]
rc选自h、卤素、羟基、nrarb、c1-c3烷基、氰基、烷氧基、=o、c3-7环烷基、3-8元杂环基、c2-c3烯基、c2-c3炔基、c(o)r
c1
、c(o)nrarb或nrac(o)rb,其中所述的烷基、环烷基、杂环烷基、烯基、ra或rb可以任选进一步被1-3个r
c2
所取代;
[0031]rc1
选自c1-c5烷基、c3-7的环烷基或3-7元的杂环基;
[0032]rc2
选自h、卤素、羟基、氨基、c1-c3烷基、烷氧基或氰基。
[0033]
r2为c1-c3烷基、c3-7的环烷基、3-8元的杂环基、6-12元的芳基或5-12元的杂芳基,其中所述的烷基、环烷基、杂环烷基、芳基或杂芳基可以任选进一步被1-5个rc所取代。
[0034]
r4选自h、卤素、c1-c3烷基、氰基或c1-2的烷氧基。
[0035]re1
、r
e2
或r
e3
分别独立选自h、卤素、c1-3的烷基、c3-7的环烷基或3-7元的杂环基,其中所述的烷基、环烷基或杂环基可以任选进一步被1-3个卤素、c1-3的烷基、羟基或氨基所取代。
[0036]
进一步具体地,所述的萘啶甲腈类衍生物具有如下表1所示的结构中的任意一种。
[0037]
表1
[0038]
[0039]
[0040][0041]
另一方面,本发明提供了上述kras突变体g12c抑制剂的制备方法,所述的制备方法包括以下步骤:
[0042][0043]
其中,
[0044]
所述的x为卤素,优选为氯。
[0045]
l1为o、s或crarb,优选为o。
[0046]
ra和rb分别独立选自h、卤素、羟基、c1-c3烷基、c3-7的环烷基或3-7元的杂环烷基,而且ra和rb连同它们所附接的原子可以进一步形成c3-7的环烷基或c3-7的杂环烷基。
[0047]
r1为6-12元的芳基或5-12元的杂芳基,所述的芳基或杂芳基可以任选进一步被1-5个rc所取代;
[0048]
rc选自h、卤素、羟基、nrarb、c1-c3烷基、氰基、烷氧基、=o、c3-7环烷基、3-8元杂环基、c2-c3烯基、c2-c3炔基、c(o)r
c1
、c(o)nrarb或nrac(o)rb,其中所述的烷基、环烷基、杂环烷基、烯基、ra或rb可以任选进一步被1-3个r
c2
所取代;
[0049]rc1
选自c1-c5烷基、c3-7的环烷基或3-7元的杂环基;
[0050]rc2
选自h、卤素、羟基、氨基、c1-c3烷基、烷氧基或氰基。
[0051]
l2为o、s或crarb,优选为o。
[0052]
r2为c1-c3烷基、c3-7的环烷基、3-8元的杂环基、6-12元的芳基或5-12元的杂芳基,其中所述的烷基、环烷基、杂环烷基、芳基或杂芳基可以任选进一步被1-5个rc所取代。
[0053]
l3选自c3-8的环烷基、c3-8螺环烷基、c3-8稠环烷基、c3-8桥环烷基、3-8元的杂环烷基、3-8元的螺杂环烷基、3-8元的稠杂环烷基、6-12元的芳基或5-12元的杂芳基,其中所述的环烷基、稠环烷基、桥环烷基、杂环基、螺杂环烷基、稠杂环烷基、芳基、杂芳基可以任选进一步被1-4个rc所取代。
[0054]
r3选自c(o)rd;
[0055]
rd选自c2-4烯基或c2-4炔基,其中所述的烯基或炔基可以任选进一步被1-3个r
d1
所取代;
[0056]rd1
选自h、卤素、羟基、nrarb、c1-c3烷基、c3-7环烷基、3-7元杂环基或5-12元的杂芳基。
[0057]
r4选自h、卤素、c1-c3烷基、氰基或c1-2的烷氧基,优选为h。
[0058]
具体地,所述的步骤(13)为通式(i-a)的化合物和通式(i-b)的化合物在碱性条件下,经取代反应得到通式(i)的化合物。
[0059]
具体地,所述的式(i-a)所示的化合物制备方法包括以下步骤:
[0060][0061]
具体地,所述的步骤(1)为通式(i-1)的化合物经氧化反应得到通式(i-2)的化合物。
[0062]
具体地,所述的步骤(2)为通式(i-2)的化合物经酯化反应得到通式(i-3)的化合物。
[0063]
具体地,所述的步骤(3)为通式(i-3)的化合物和通式(i-4)的化合物在碱性条件下经取代反应得到通式(i-5)的化合物。
[0064]
具体地,所述的步骤(4)为通式(i-5)的化合物在碱性条件下,上甲基得到通式(i-6)的化合物。
[0065]
具体地,所述的步骤(5)为通式(i-6)的化合物经还原反应得到通式(i-7)的化合物。
[0066]
具体地,所述的步骤(6)为通式(i-7)的化合物在碱性条件下和氰基乙酸经缩合反应得到通式(i-8)的化合物。
[0067]
具体地,所述的步骤(7)为通式(i-8)的化合物在碱性条件下,关环得到通式(i-9)
的化合物。
[0068]
具体地,所述的步骤(8)为通式(i-9)的化合物在碱性条件下,羟基卤代得到通式(i-10)的化合物。
[0069]
具体地,所述的步骤(9)为通式(i-10)的化合物和通式(i-11)的化合物在碱性条件下,经取代反应得到通式(i-12)的化合物。
[0070]
具体地,所述的步骤(10)为通式(i-12)的化合物在碱性条件下,和三氟甲烷磺酸酐反应得到通式(i-13)的化合物。
[0071]
具体地,所述的步骤(11)为通式(i-13)的化合物和通式(i-14)的化合物在碱性条件下,经取代反应得到通式(i-15)的化合物。
[0072]
进一步具体地,所述的通式(i-15)的化合物还能通过以下方法制备得到:通式(i-12)的化合物和通式(i-14)的化合物经mitsunobu反应得到通式(i-15)的化合物。
[0073]
具体地,所述的步骤(12)为通式(i-15)的化合物在酸性条件下脱保护基得到通式(i-a)的化合物。
[0074]
进一步具体地,上述步骤中提供碱性条件的试剂选自有机碱或无机碱,所述的有机碱为三乙胺、n,n-二异丙基乙胺、正丁基锂、二异丙基氨基锂、双三甲基硅基胺基锂、叔丁醇钠、甲醇钠或叔丁醇钾中的一种或多种,所述的无机碱为氢化钠、磷酸钾、碳酸钠、碳酸钾、醋酸钾、碳酸铯、氢氧化钠、氢氧化钾、碳酸氢钠或氢氧化锂中的一种或多种。
[0075]
进一步具体地,上述步骤中提供酸性条件的试剂为氯化氢、氯化氢的1,4-二氧六环溶液、氯化氢的甲醇溶液、三氟乙酸、甲酸、乙酸、盐酸、硫酸、甲磺酸、硝酸或磷酸中的一种或多种。
[0076]
进一步具体地,上述步骤中金属催化剂为钯/碳、雷尼镍、四-三苯基膦钯、二氯化钯、醋酸钯、[1,1'-双(二苯基膦基)二茂铁]二氯化钯(pd(dppf)cl2)、[1,1'-双(二苯基膦基)二茂铁]二氯化钯二氯甲烷络合物、双三苯基磷二氯化钯(pd(pph3)cl2)或三(二亚苄基丙酮)二钯(pd2(dba)3)中的一种或多种。
[0077]
进一步具体地,上述步骤中配体为2-双环己基膦-2,6'-二甲氧基联苯(sphos)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(xantphos)、2-二环己基磷-2,4,6-三异丙基联苯(xphos)、2-二环己膦基-2'-(n,n-二甲胺)-联苯(davephos)、1,1'-双(二苯基膦)二茂铁(dppf)和1,1'-联萘-2,2'-双二苯膦(binap)中的一种或多种,优选为1,1'-联萘-2,2'-双二苯膦(binap)。
[0078]
进一步具体地,上述步骤中还原剂为硼氢化钠、硼氢化钾、氰基硼氢化钠、三乙酰氧基硼氢化钠或四氢铝锂中的一种或多种。
[0079]
进一步具体地,上述步骤中氧化剂为高锰酸钾、二氧化锰、重铬酸钾、重铬酸钠或锇酸钾中的一种或多种。
[0080]
进一步具体地,上述步骤优选在溶剂中进行,所用溶剂为n,n-二甲基甲酰胺、n-甲基吡咯烷酮、二甲基亚砜、1,4-二氧六环、水、四氢呋喃、二氯甲烷、1,2-二氯乙烷、甲醇、乙醇、甲苯、石油醚、乙酸乙酯、正己烷或丙酮中的一种或多种。
[0081]
又一方面,本发明提供了上述kras突变体g12c抑制剂在制备药物组合物中的应用。
[0082]
又一方面,本发明提供了一种药物组合物,所述的药物组合物含有上述治疗有效
量的kras突变体g12c抑制剂,以及余量的药学上可接受的载体和/或赋形剂。
[0083]
又一方面,本发明提供了上述kras突变体g12c抑制剂或药物组合物在制备用于治疗通过抑制ras通路可治疗的疾病的药物中的应用。
[0084]
具体地,所述的疾病为癌症。
[0085]
又一方面,本发明提供了上述kras突变体g12c抑制剂或药物组合物在制备用于治疗癌症的药物中的应用。
[0086]
具体地,所述癌症为肺癌、胰腺癌或结直肠癌。
[0087]
与现有技术相比,本发明的积极和有益效果在于:
[0088]
本发明提供了一种新的kras突变体g12c抑制剂,所述的kras突变体g12c抑制剂为萘啶甲腈类衍生物、其药学上可接受的盐、其互变异构体或其立体异构体,试验结果表明,该萘啶甲腈类衍生物表现出优异的kras突变体g12c抑制活性,其具有较好的抗肿瘤有效性、稳定性、选择性及安全性,具有广泛的抗肿瘤药物应用前景,尤其是肺癌、结直肠癌或胰腺癌等疾病。
附图说明
[0089]
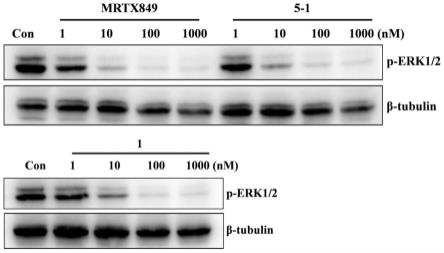
图1为本发明实施例2-8制备的化合物对miapaca-2细胞中kras/erk1/2信号转导通路的检测结果图。
具体实施方式
[0090]
下面结合具体实施例,对本发明作进一步详细的阐述,下述实施例不用于限制本发明,仅用于说明本发明。以下实施例中所使用的实验方法如无特殊说明,实施例中未注明具体条件的实验方法,通常按照常规条件,下述实施例中所使用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0091]
1、除非有相反陈述,否则本发明在说明书和权利要求书中所使用的部分术语定义如下:
[0092]“烷基”指饱和脂肪族烃基团,包括1-20个碳原子,或1-10个碳原子,或1-6个碳原子,或1-4个碳原子,或1-3个碳原子,或1-2个碳原子饱和直链或支链的单价烃基,其中烷基可以独立任选地被一个或多个本发明所描述地取代基所取代。烷基基团更近一步地实例包括,包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、1,1-二甲基丙基、1,2-二甲基丙基、2,2-二甲基丙基、1-乙基丙基、2-甲基丁基、3-甲基丁基、正己基、1-乙基-2-甲基丙基、1,1,2-三甲基丙基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1,3-二甲基丁基、2-乙基丁基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2,3-二甲基丁基等。烷基可以是任选取代或未取代的。
[0093]“烯基”指2-12个碳原子,或2-8个碳原子,或2-6个碳原子,或2-4个碳原子直链或支链的一价烃基,其中至少一个c-c为sp2双键,其中烯基的基团可以独立任选地被1个或多个本发明所描述的取代基所取代,其中具体的实例包括,但并不限于乙烯基、烯丙基和烯丁基等等。烯基可以是任选取代或未取代的。
[0094]“环烷基”是指饱和或部分不饱和单环或多环环状烃取代基,环烷基环包括3至20个碳原子,优选包括3至12个碳原子,更优选包含3至6个碳原子。单环环烷基的非限制性实
施例包括,但不限于环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环己二烯基、环庚基、环庚三烯基、环辛基等;多环环烷基包括螺环、稠环和桥环的环烷基。环烷基可以是任选取代的或未取代的。
[0095]“螺环烷基”指5至18元,两个或两个以上环状结构,且单环之间彼此共用一个碳原子(称螺原子)的多环基团,环内含有1个或多个双键,但没有一个环具有完全共轭的π电子的芳香系统。优选为6至14元,更优选为7至10元。根据环与环之间共用螺原子的数目将螺环烷基分为单螺、双螺或多螺环烷基,优选为单螺和双螺环烷基,优选为4元/5元、4元/6元、5元/5元或5元/6元。“螺环烷基”的非限制性实施例包括但不限于:
[0096][0097]“稠环烷基”指5至18元,含有两个或两个以上环状结构彼此公用一对碳原子的全碳多环基团,一个或多个环可以含有一个或多个双键,但没有一个环具有完全共轭的π电子的芳香系统,优选为6至12元,更优选为7至10元。根据组成环的数目可以分为双环、三环、四环或多环稠环烷基,优选为双环或三环,更优选为5元/5元或5元/6元双环烷基。“稠环烷基”的非限制性实施例包括但不限于:
[0098][0099]“桥环烷基”指5至18元,含有两个或两个以上环状结构,彼此共用两个不直接相连接碳原子的全碳多环基团,一个或多个环可以含有一个或多个双键,但没有一个环具有完全共轭的π电子的芳香系统,优选为6至12元,更优选为7至10元。根据组成环的数目可以分为双环、三环、四环或多环桥环烷基,优选为双环、三环或四环,更有选为双环或三环。“桥环烷基”的非限制性实施例包括但不限于:
[0100][0101]
所述环烷基环可以稠合于芳基、杂芳基或杂环基环上,其中与母体结构连接在一起的环为环烷基,非限制性实施例包括茚满基、四氢萘基、苯并环庚烷基等。
[0102]“杂环基”、“杂环”或“杂环的”在本技术中可交换使用,本技术中可交换使用,都是指包含3-12个环原子的饱和或部分不饱和的单环、双环或三环的非芳香性杂环基,其中至少一个环原子原子是杂原子,如氧、氮、硫原子等。优选具有5至7元单环或7至10元双-或三环,其可以包含1,2或3个选自氮、氧和/或硫中的原子。“杂环基”的实例包括但不限于吗啉基,氧杂环丁烷基,硫代吗啉基,四氢吡喃基,1,1-二氧代-硫代吗啉基,哌啶基,2-氧代-哌啶基,吡咯烷基,2-氧代-吡咯烷基,哌嗪-2-酮,8-氧杂-3-氮杂-双环[3.2.1]辛基和哌嗪基。所述杂环基环可以稠合于芳基、杂芳基或环烷基环上,其中与母体结构连接在一起的环为杂环基。杂环基可以是任选取代的或未取代的。
[0103]“螺杂环基”指5至18元,两个或两个以上环状结构,且单环之间彼此共用一个原子的多环基团,环内含有1个或多个双键,但没有一个环具有完全共轭的π电子的芳香系统,其中一个或多个环原子选自氮、氧、硫或s(o)m的杂原子,其余环原子为碳,m=1或2。优选为6至14元,更优选为7至10元。根据环与环之间共用螺原子的数目将螺杂环基分为单螺杂环基、双螺杂环基或多螺杂环基,优选为单螺杂环基和双螺杂环基。更优选为4元/4元、4元/5元、4元/6元、5元/5元或5元/6元单螺杂环基。“螺杂环基”的非限制性实施例包括但不限于:
[0104][0105]“稠杂环基”指含有两个或两个以上环状结构彼此公用一对原子的全碳多环基团,一个或多个环可以含有一个或多个双键,但没有一个环具有完全共轭的π电子的芳香系统,其中一个或多个环原子选自氮、氧、硫或s(o)m的杂原子,其余环原子为碳,m=1或2。优选为6至14元,更优选为7至10元。根据组成环的数目可以分为双环、三环、四环或多环稠杂环基,优选为双环或三环,更优选为5元/5元或5元/6元双环稠杂环基。“稠杂环基”的非限制性实施例包括但不限于:
[0106][0107]“桥杂环基”指5至18元,含有两个或两个以上环状结构,彼此共用两个不直接相连接的原子的多环基团,一个或多个环可以含有一个或多个双键,但没有一个环具有完全共轭的π电子的芳香系统,其中一个或多个环原子选自氮、氧、硫或s(o)m的杂原子,其余环原子为碳,m=1或2。优选为6至14元,更优选为7至10元。根据组成环的数目可以分为双环、三环、四环或多环桥杂环基,优选为双环、三环或四环,更有选为双环或三环。“桥杂环基”的非限制性实施例包括但不限于:
[0108][0109]“芳基”是指含有一个或者两个环的碳环芳香系统,其中所述环可以以稠合的方式连接在一起。术语“芳基”包括比如苯基、萘基、四氢萘基的芳香基团。优选芳基为c
6-c
10
芳基,更优选芳基为苯基和萘基,最优选为苯基。芳基可以是取代或未取代的。所述“芳基”可与杂芳基、杂环基或环烷基稠合,其中与母体结构连接在一起的为芳基环,非限制性实施例包括但不限于:
[0110][0111]“杂芳基”是指芳香族5至6元单环或9至10元双环,其可以包含1至4个选自氮、氧和/或硫中的原子。“杂芳基”的实施例包括但不限于呋喃基,吡啶基,2-氧代-1,2-二氢吡啶基、哒嗪基、嘧啶基、吡嗪基、噻吩基、异噁唑基、噁唑基、噁二唑基、咪唑基、吡咯基、吡唑基、三唑基、四唑基、噻唑基、异噻唑基、1,2,3-噻二唑基、苯并间二氧杂环戊烯基、苯并咪唑基、吲哚基、异吲哚基、1,3-二氧代-异吲哚基、喹啉基、吲唑基、苯并异噻唑基、苯并噁唑基和苯并异噁唑基。杂芳基可以是任选取代或未取代的。所述杂芳基环可以稠合于芳基、杂环基或环烷基环上,其中与母体结构连接在一起的环为杂芳基环,非限制性实施例包括但不限于:
[0112][0113]“烷氧基”是指(烷基-o-)的基团。其中,烷基见本文有关定义。c
1-c6的烷氧基为优先选择。其实例包括,但不限于:甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基等。
[0114]“卤代烷基”指具有一个或者多个卤素取代基的烷基,其中烷基基团具有如本发明所述的含义。卤代烷基的实例包括,但并不限于氟甲基、二氟甲基、三氟甲基、全氟乙基、1,1-二氯乙基、1,2-二氯丙基等。
[0115]“羟基”指-oh基团。
[0116]“卤素”是指氟、氯、溴和碘,优选氟、氯和溴。
[0117]“氨基”指-nh2。
[0118]“氰基”指-cn。
[0119]“硝基”指-no2。
[0120]“苄基”指-ch
2-苯基。
[0121]“羧基”指-c(o)oh。
[0122]“乙酰基”指-c(o)ch3或ac。
[0123]“羧酸酯基”指-c(o)o(烷基)或(环烷基),其中烷基、环烷基的定义如上所述。
[0124]“任选”意味着其所描述的事件可以但不必发生。例如,“ar1任选地被1到多个rc取代”该说明包含着ar1基团可以被1到多个rc取代或者不被rc取代的情形。
[0125]“取代的”指基团中的一个或多个氢原子,优选为最多5个,更优选为1-3个氢原子彼此独立地被相应数目的取代基取代。不言而喻,取代基仅处在它们的可能的化学位置,本
领域技术人员能够在不付出过多努力的情况下确定(通过实验或理论)可能或不可能的取代。例如,具有游离氢的氨基或羟基与具有不饱和(如烯属)键的碳原子结合时可能是不稳定的。
[0126]
本说明书所述的“取代”或“取代的”,如无特别指出,均是指基团可被一个或多个选自以下的基团取代:烷基、烯基、炔基、烷氧基、烷硫基、烷基氨基、卤素、疏基、羟基、硝基、氰基、环烷基、杂环基、芳基、杂芳基、环烷氧基、杂环烷氧基、环烷硫基、杂环烷硫基、氨基、卤代烷基、羟烷基、羧基、羧酸酯基、=o、-c(o)rb、-oc(o)rb、-nrbrb、-c(o)nrbrb、-nrbc(o)rb、-s(o)nrbrb或-s(o)2nrbrb,其中,rb的定义如通式(i)中所述。
[0127]
本发明中立体化学的定义和惯例的使用通常参考以下文献:s.p.parker,ed.,mcgraw-hill dictionary of chemicalterms(1984)mcgraw-hillbookcompany,newyork;andeliel,e.andwilen,s.,"stereochemistry of organic compounds",john wiley&sons,inc.,new york,1994。
[0128]
本发明的化合物可以包含不对称中心或手性中心,因此存在不同的立体异构体。本发明的化合物所有的立体异构形式,包括但绝不限于,非对映体,对映异构体,阻转异构体,和它们的混合物,如外消旋混合物,组成了本发明的一部分。非对映异构体可以以其物理化学差异为基础,通过层析、结晶、蒸馏或升华等方法被分离为个别非对映异构体。对映异构体可以通过分离,使手性异构混合物转化为非对映异构混合物,其方式是与适当光学活性化合物(例如手性辅助剂,譬如手性醇或mosher氏酰氯)的反应,分离非对映异构体,且使个别非对映异构体转化为相应的纯对映异构体。本发明的中间体与化合物也可以不同互变异构形式存在,且所有此种形式被包含在本发明的范围内。很多有机化合物都以光学活性形式存在,即它们有能力旋转平面偏振光的平面。在描述光学活性化合物时,前缀d、l或r、s用来表示分子手性中心的绝对构型。前缀d、l或( )、(-)用来命名化合物平面偏振光旋转的符号,(-)或l是指化合物是左旋的,前缀( )或d是指化合物是右旋的。这些立体异构体的原子或原子团互相连接次序相同,但是它们的立体结构不一样。特定的立体异构体可以是对映体,异构体的混合物通常称为对映异构体混合物。50:50的对映体混合物被称为外消旋混合物或外消旋体,这可能导致化学反应过程中没有立体选择性或立体定向性。术语“外消旋混合物”和“外消旋体”是指等摩尔的两个对映异构体的混合物,缺乏光学活性。
[0129]“互变异构体”或“互变异构的形式”是指不同能量的结构的同分异构体可以通过低能垒互相转化。例如质子互变异构体(即质子移变的互变异构体)包括通过质子迁移的互变,如酮式-烯醇式和亚胺-烯胺的同分异构化作用。原子价(化合价)互变异构体包括重组成键电子的互变。除非其他方面表明,本发明所描述的结构式包括所有的同分异构形式(如对映异构,非对映异构,和几何异构):例如含有不对称中心的r、s构型,双键的(z)、(e)异构体,和(z)、(e)的构象异构体。因此,本发明的化合物的单个立体化学异构体或其对映异构体,非对映异构体,或几何异构体的混合物都属于本发明的范围。
[0130]“可药用盐”指本发明化合物的盐,这类盐用于人或动物体内时具有安全性和有效性。化合物的盐可以通过在纯的溶液或合适的惰性溶解中用足量的碱或酸获得相应的加成盐。可药用的碱加成盐包括钠、钾、钙、铵、有机氨或镁盐等,可药用的酸加成盐包括无机酸盐和有机酸盐,所述的无机酸和有机酸包括盐酸、氢溴酸、碳酸、碳酸氢根、磷酸、磷酸一氢根、磷酸二氢根、硫酸、硫酸一氢根、乙酸、马来酸、丙二酸、琥珀酸、饭丁烯二酸、邻苯二甲
酸、苯磺酸、对甲苯磺酸、柠檬酸和甲磺酸等(参见berge et al.,“pharmaceutical salts”,journal of pharmaceutical science 66:1-19(1977))。
[0131]
2、实验仪器:
[0132]
下述实施例中1h nmr图谱是用bruker仪器(400mhz)测定而得,化学位移用ppm表示。使用四甲基硅烷内标准(0.00ppm)。1h nmr的表示方法:s=单峰,d=双重峰,t=三重峰,q=四重峰,m=多重峰,br=变宽的,dd=双重峰的双重峰,dt=三重峰的双重峰。若提供偶合常数时,其单位为hz。
[0133]
质谱是用lc/ms仪测定得到,离子化方式为esi。
[0134]
高效液相色谱仪型号:安捷伦1260、赛默飞u3000;色谱柱型号:waters xbrige c18(4.6*150mm,3.5μm);流动相:a:acn,b:water(0.1%h3po4);流速:
[0135]
1.0ml/min;梯度:5%a for 1min,increase to 20%a within 4min,increase to 80%a within 8min,80%a for 2min,back to 5%a within 0.1min;波长:220nm;柱温箱:35℃。
[0136]
薄层层析硅胶板使用烟台黄海hsgf254或青岛gf254硅胶板,薄层色谱法(tlc)使用的硅胶板采用的规格是0.2-0.3mm,薄层层析分离纯化产品采用的规格是0.4-0.5mm。
[0137]
柱层析一般使用烟台黄海硅胶200-300目硅胶为载体。
[0138]
tlc:薄层色谱法。
[0139]
hplc:高效液相色谱法。
[0140]
3、实验试剂:
[0141]
在下列实例中,除非另有指明,所有温度为摄氏温度,除非另有指明,各种起始原料和试剂来自市售或者是根据已知的方法合成,市售原料和试剂均不经进一步纯化直接使用,除非另有指明,市售厂家包括但不限于国药集团,百灵威科技有限公司,梯希爱(上海)化成工业发展有限公司,上海毕得医药科技有限公司和上海迈瑞尔化学科技有限公司等。
[0142]
cd3od:氘代甲醇。
[0143]
cdcl3:氘代氯仿。
[0144]
dmso-d6:氘代二甲基亚砜。
[0145]
pd2(dba)3:三(二亚苄基丙酮)二钯。
[0146]
t-buxphos:2-二-叔丁膦基-2',4',6'-三异丙基联苯。
[0147]
purity:纯度。
[0148]
&:和。
[0149]
氢气氛围是指反应瓶连接一个约1l容积的氢气气球。
[0150]
实施例中无特殊说明,反应中的溶液是指水溶液。
[0151]
实施例中无特殊说明,反应的温度为室温,为20-30℃。
[0152]
实施例中的反应进程的监测采用薄层色谱法(tlc),反应所使用的展开剂,纯化化合物采用的柱层析的洗脱剂的体系或薄层色谱法的展开剂体系包括:a:石油醚和乙酸乙酯体系;b:二氯甲烷和甲醇体系;c:正己烷:乙酸乙酯;其中溶剂的体积比根据化合物的极性不同而不同,也可以加入少量的酸性或碱性试剂进行调节,如醋酸或三乙胺等。
[0153]
实施例1.中间体的制备
[0154]
1、中间体1
[0155]
(1)in-1-1:4-氯-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-氧代-1,2-二氢-1,7-萘啶-3-甲腈。
[0156]
(2)in-1-2:4-氯-8-((5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑-4-基)氧基)-2-氧代-1,2-二氢-1,7-萘啶-3-甲腈。
[0157][0158]
2、制备方法:
[0159][0160]
步骤(1):3-溴-5-氟-2-甲基苯胺in-1b
[0161]
2-溴-4-氟-6-硝基甲苯in-1a(100g,427.3mmol)分散于乙醇(1l)和水(500ml)中,升温至70℃,分批加入氯化铵(143g,2.67mol)和铁粉(140g,2.51mol),加完70℃反应5小时,tlc显示反应完全。反应液趁热过滤,滤饼乙酸乙酯洗涤,滤液乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得无色油状液体标题化合物in-1b(96.1g,粗品),直接用于下一步。
[0162]
lc-ms:m/z=204.0[m h]
。
[0163]
步骤(2):3-溴-4-氯-5-氟-2-甲基苯胺in-1c
[0164]
化合物in-1b(96.1g,粗品)分散于异丙醇(1l)中,升温至40℃,分批加入n-氯代丁二酰亚胺(68.5g,512.8mmol)),加完40℃搅拌4小时,tlc显示原料基本反应完全。反应液冷却至室温,加水,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析分离纯化得砖红色固体化合物in-1c(58.0g,两步收率62%)。
[0165]
lc-ms:m/z=239.9[m h]
。
[0166]
步骤(3):4-溴-5-氯-6-氟-1h-吲唑in-1d
[0167]
化合物in-1c(58.0g,243.2mmol)分散于醋酸中(620ml)中,冷却至0℃,分批加入亚硝酸钠(67.1g,972.5mmol),加完升至室温搅拌15小时,tlc显示反应完全。反应液加水淬
灭,乙酸乙酯萃取,合并有机相,水洗五次,饱和碳酸氢钠溶液洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析分离纯化得砖红色固体化合物in-1d(41.0g,收率68%)。
[0168]
lc-ms:m/z=251.0[m h]
。
[0169]
步骤(4):4-溴-5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑in-1e
[0170]
化合物in-1d(41.0g,164.3mmol)分散于乙腈(600ml)中,室温下加入对甲苯磺酸一水合物(3.2g,16.4mmol)和3,4-二氢-2h-吡喃(41.5g,493.0mmol),升温至30℃搅拌2小时,tlc显示反应完全。反应液加水淬灭,饱和碳酸钠溶液调ph至8-9,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析分离纯化得砖红色固体化合物in-1e(37.5g,收率68%)。
[0171]
步骤(5):5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑-4-醇in-1f
[0172]
化合物in-1e(37.5g,112.4mmol)分散于1,4二氧六环(400ml)中,室温下加入氢氧化钾(18.9g,336.9mmol)的水(100ml)溶液,室温下依次加入t-buxphos(2.5g,5.89mmol)和pd2(dba)3(2.5g,2.73mmol),氮气保护下,升温至95℃反应3小时,tlc显示反应完全。反应液降至室温,加水稀释,乙酸乙酯萃取,丢弃有机相,水相用稀盐酸(1n)调ph至4-5,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析分离纯化得红色固体化合物in-1f(26.8g,收率88%)。
[0173]
lc-ms:m/z=271.1[m h]
。
[0174]
步骤(6):2-氯-3-硝基异烟酸in-1h
[0175]
2-氯-4-甲基-3-硝基吡啶in-1g(20.0g,115.9mmol)溶于浓硫酸(200ml)中,室温下缓慢加入重铬酸钾(45.9g,156.0mmol),加毕升温至至60℃反应3小时,tlc显示反应完全。反应液冷却至室温,缓慢倒入冰水中,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得白色固体化合物in-1h(20.1g,粗品),直接用于下一步。
[0176]
lc-ms:m/z=201.0[m-h]-。
[0177]
步骤(7):2-氯-3-硝基异烟酸甲酯in-1i
[0178]
化合物in-1h(15.0g,粗品)溶于甲醇(150ml)中,室温下缓慢滴加氯化亚砜(12.9g,109.4mmol),滴毕加热至回流反应12小时,tlc显示反应完全。反应液冷却至室温,浓缩,倒入冰水中,饱和碳酸氢钠调节ph至8-9,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得浅黄色固体化合物in-1i(15.1g,粗品),直接用于下一步。
[0179]
lc-ms:m/z=217.1[m h]
。
[0180]
步骤(8):2-((5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑-4-基)氧基)-3-硝基异烟酸in-1j
[0181]
化合物in-1i(2.2g,粗品)溶于二甲基亚砜(30ml)中,室温下加入中间体in-1(2.5g,9.24mmol))和碳酸铯(6.0g,18.42mmol)),升温至100℃反应1小时,tlc显示反应完。反应液冷却至室温,缓慢倒入冰水中,4m盐酸水溶液调节ph至2-3,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得黑色油状物化合物in-1j(5.8g,粗品),直接用于下一步。
[0182]
步骤(9):2-((5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑-4-基)氧基)-3-硝基异烟酸甲酯in-1k
[0183]
化合物in-1j(5.8g,粗品)和碳酸钠(1.2g,11.32mmol)分散至n,n-二甲基甲酰胺
(20ml)中,冷却至0℃,加入碘甲烷(987mg,6.95mmol),升至室温反应2小时,tlc显示原料剩余少量。反应液倒入冰水中,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得浅黄色固体化合物in-1k(2.7g,四步收率45%)。
[0184]
lc-ms:m/z=451.1[m h]
。
[0185]
步骤(10):3-氨基-2-((5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑-4-基)氧基)异烟酸甲酯in-1l
[0186]
化合物in-1k(2.7g,5.99mmol)溶于醋酸(50ml)中,室温下加入铁粉(10.0g,179.0mmol),升温至60℃反应3小时,tlc显示反应完。反应液降至室温,浓缩除去醋酸,饱和碳酸氢钠溶液调节ph至8-9,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得浅黄色固体化合物in-1l(2.5g,收率99%)。
[0187]
步骤(11):2-((5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑-4-基)氧基)-3-(2-氰基乙酰胺基)异烟酸甲酯in-1m
[0188]
化合物in-1l(2.5g,5.94mmol)溶于乙酸乙酯(30ml)中,室温下依次加入吡啶(1.9g,24.02mmol),1-丙基磷酸酐(7.6g,119.4mmol,50%w/w乙酸乙酯溶液)和氰基乙酸(612mg,7.19mmol),升温至80℃反应2小时,tlc显示反应完。反应液降至室温,倒入水中,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得黄色固体化合物in-1m(2.3g,粗品),直接下一步。
[0189]
lc-ms:m/z=488.1[m h]
。
[0190]
步骤(12):8-((5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑-4-基)氧基)-4-羟基-2-氧代-1,2-二氢-1,7-萘啶-3-甲腈in-1n
[0191]
化合物in-1m(2.3g,粗品)溶于四氢呋喃(30ml)中,降温至0-5℃,加入氢化钠(284mg,7.10mmol,60%),升至室温反应1小时,tlc显示反应完全。反应液缓慢倒入冰水(5ml)中,4m盐酸调节ph至2-3,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得深黄色固体标题化合物in-1n(2.2g,粗品),直接用于下一步。
[0192]
lc-ms:m/z=456.1[m h]
。
[0193]
步骤(13):4-氯-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-氧-1,2-二氢-1,7-萘啶-3-腈in-1-1&4-氯-8-((5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑-4-基)氧基)-2-氧-1,2-二氢-1,7-萘啶-3-甲腈in-1-2
[0194]
化合物in-1n(1.8g,粗品)溶于三氯氧磷(11ml)中,室温下,恒压滴液漏斗缓缓滴入n,n-二异丙基乙胺(1.2g,9.2mmol)和n,n-二甲基甲酰胺(1ml),加完室温反应20分钟,tlc显示反应完全。反应液缓慢倒入热的饱和碳酸氢钠溶液中(100ml),乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,缩干,粗品硅胶柱层析纯化得黄色固体化合物in-1-1(900mg,三步收率49%)和黄色固体化合物in-1-2(550mg,三步收率20%)。
[0195]
化合物in-1-1:
[0196]
lc-ms:m/z=388.0[m-h]-。
[0197]1h nmr(400mhz,dmso-d6)δ13.58(s,1h),13.16(s,1h),7.92-7.89(m,2h),7.65-7.61(m,2h)。
[0198]
化合物in-1-2:
[0199]
lc-ms:m/z=472.0[m-h]-。
[0200]1h nmr(400mhz,dmso-d6)δ13.19(br 1h),8.00(s 1h),7.96-7.87(m,2h),7.70-7.63(m,1h),5.92-5.84(m,1h),3.96-3.87(m,1h),3.84-3.74(m,1h),2.41-2.31(m,1h),2.09-1.94(m,2h),1.82-1.65(m,1h),1.65-1.53(m,2h)。
[0201]
实施例2
[0202]
4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2-甲基-1,2,3,4-四氢异喹啉-5-基)氧基)-1,7-萘啶-3-甲腈(式1化合物)
[0203][0204]
制备方法:
[0205][0206]
步骤(1):5-羟基-2-甲基异喹啉-2-盐1b
[0207]
5-羟基异喹啉1a(9.5g,65.45mmol)溶于无水甲醇(200ml)中,室温下加入碘甲烷(46.45g,327.25mmol),升温至45℃搅拌24小时,tlc显示原料剩余少量。反应液降至室温,浓缩得固体化合物1b(12.8g,粗品),直接用于下一步。
[0208]
步骤(2):5-羟基-2-甲基-1,2,3,4-四氢异喹1c
[0209]
化合物1b(12.8g,79.90mmol)溶于无水甲醇(200ml)中,降温至0℃,分批加入硼氢化钠(13.6g,359.50mmol),加完室温搅拌1小时,tlc显示反应完。反应液加水淬灭,浓缩去除甲醇,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得黄色固体化合物1c(9.0g,两步收率84%)。
[0210]1h nmr(400mhz,dmso-d6)δ9.28(s,1h),6.91(t,j=7.8hz,1h),6.61(d,j=7.8hz,
1h),6.48(d,j=7.8hz,1h),3.41(s,2h),2.62-2.60(m,2h),2.58-2.56(m,2h),2.32(s,3h)。
[0211]
步骤(3):4-(8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-3-氰基-2-羟基-1,7-萘啶-4-基)哌嗪-1-甲酸叔丁酯1d
[0212]
实施例1制备得到的中间体in-1-1(700mg,1.79mmol)溶于n,n-二甲基甲酰胺(20ml)中,依次加入n,n-二异丙基乙胺(692mg,5.35mmol)和1-boc-哌嗪(500mg,2.70mmol),室温反应2小时,tlc显示反应完全。反应液加水稀释,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥浓缩,粗品硅胶柱层析纯化得黄色固体化合物1d(280mg,收率29%)。
[0213]
lc-ms:m/z=538.2[m-h]-。
[0214]
步骤(4):4-((4-(4-(叔丁氧基羰基)哌嗪-1-基)-3-氰基-2-羟基-1,7-萘啶-8-基)氧基)-5-氯-6-氟-1h-吲唑-1-甲酸叔丁酯1e
[0215]
化合物1d(280mg,0.52mmol)溶于二氯甲烷(40ml),加入二碳酸二叔丁酯(169mg,0.77mmol)和4-二甲氨基吡啶(6mg,0.05mmol),室温反应过夜,tlc显示原料反应完。反应液加入二氯甲烷稀释,水洗,饱和食盐水洗,无水硫酸钠干燥,浓缩得化合物1e(295mg,粗品),直接用于下一步。
[0216]
步骤(5):4-((4-(4-(叔丁氧基羰基)哌嗪-1-基)-3-氰基-2-(((三氟甲基)磺酰基)氧基)-1,7-萘啶-8-基)氧基)-5-氯-6-氟-1h-吲唑-1-甲酸叔丁酯1f
[0217]
化合物1e(295mg,粗品)和三乙胺(186mg,1.84mmol)溶于二氯甲烷(20ml)中,室温下滴加三氟甲烷磺酸酐(195mg,0.69mmol),加完室温反应10分钟,tlc显示原料剩余少量。反应液加入二氯甲烷稀释,水洗,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得固体化合物1f(175mg,两步收率44%)。
[0218]
步骤(6):4-(8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-3-氰基-2-((2-甲基-1,2,3,4-四氢异喹啉-5-基)氧基)-1,7-萘啶-4-基)哌嗪-1-甲酸叔丁酯1g
[0219]
化合物1f(175mg,0.23mmol)溶于n,n-二甲基甲酰胺(20ml),室温下加入碳酸铯(220mg,0.68mmol)和化合物1c(55mg,0.34mmol),升温至40℃反应1小时,tlc显示反应完。反应液降至室温,加入乙酸乙酯稀释,水洗,饱和食盐水洗,无水硫酸钠干燥,浓缩得黄色固体化合物1g(195mg,粗品),直接用于下一步。
[0220]
lc-ms:m/z=685.2[m h]
。
[0221]
步骤(7):8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2-甲基-1,2,3,4-四氢异喹啉-5-基)氧基)-4-(哌嗪-1-基)-1,7-萘啶-3-甲腈1h
[0222]
化合物1g(195mg,粗品)溶于二氯甲烷(2.5ml)中,加入三氟乙酸(5ml),室温搅拌3小时,tlc显示反应完全。反应液加入二氯甲烷稀释,水洗,饱和碳酸钠溶液洗至碱性,饱和食盐水洗,无水硫酸钠干燥,浓缩得黄色固体化合物1h(120mg,粗品),直接用于下一步。
[0223]
lc-ms:m/z=585.2[m h]
。
[0224]
步骤(8):4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2-甲基-1,2,3,4-四氢异喹啉-5-基)氧基)-1,7-萘啶-3-甲腈(式1化合物)
[0225]
化合物1h(120mg,粗品)溶于二氯甲烷(10ml)中,加入水(10ml)和碳酸氢钠(69mg,0.82mmol),搅拌下,慢慢滴加丙烯酰氯(18mg,0.205mmol)的二氯甲烷(1ml)溶液,滴完室温
搅拌1分钟,tlc显示反应完全。反应液加入二氯甲烷稀释,水洗,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品prep-hplc纯化得白色固体化合物1(10mg,三步收率7%)。
[0226]
lc-ms:m/z=639.2[m h]
。
[0227]1h nmr(400mhz,dmso-d6)δ13.49(s,1h),7.93(d,j=6.0hz,1h),7.66(d,j=6.0hz,1h),7.62(s,1h),7.51(d,j=8.8hz,1h),7.19-7.15(m,1h),7.12-7.10(m,1h),6.97-6.89(m,2h),6.21(dd,j=16.4,2.0hz,1h),5.77(dd,j=9.6,2.0hz,1h),3.91-3.82(m,8h),3.50(s,2h),2.65-2.63(m,2h),2.58-2.55(m,2h),2.31(s,3h),(96.55%purity by hplc)。
[0228]
实施例3
[0229]
(s)-4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((1-甲基吡咯烷-2-基)甲氧基)-1,7-萘啶-3-甲腈(式2化合物)
[0230][0231]
制备方法:
[0232][0233]
步骤(1):4-(8-((5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑-4-基)氧基)-3-氰基-2-羟基-1,7-萘啶-4-基)哌嗪-1-甲酸叔丁酯2a
[0234]
中间体in-1-2(740mg,1.56mmol)溶于n,n-二甲基甲酰胺(5ml)中,加入三乙胺(474mg,4.68mmol)和1-boc-哌嗪(320mg,1.72mmol),室温反应1小时,tlc显示反应完全。反应液加水稀释,乙酸乙酯萃取,合并有机相,半饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品
硅胶柱层析纯化得化合物2a(950mg,收率97%)。
[0235]
步骤(2):4-(8-((5-氯-6-氟-1-(四氢-2h-吡喃-2-基)-1h-吲唑-4-基)氧基)-3-氰基-2-(((s)-1-甲基吡咯烷-2-基)甲氧基)-1,7-萘啶-4-基)哌嗪-1-甲酸叔丁酯2b
[0236]
化合物2a(100mg,0.16mmol),n-甲基-l-脯氨醇(37mg,0.32mmol)和三苯基膦(84mg,0.32mmol)置于三口瓶中,氮气保护下,加入干燥四氢呋喃(4ml),冷却至0℃,加入偶氮二甲酸二异丙酯(65mg,0.32mmol),室温反应过夜,tlc显示反应完全。反应液加入二氯甲烷和水,萃取分液,有机相无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得化合物2b(80mg,收率69%)。
[0237]
lc-ms:m/z=721.3[m h]
。
[0238]
步骤(3):(s)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((1-甲基吡咯烷-2-基)甲氧基)-4-(哌嗪-1-基)-1,7-萘啶-3-甲腈2c
[0239]
化合物2b(80mg,0.11mmol)溶于二氯甲烷(1ml)中,加入三氟乙酸(1ml),室温反应1小时,tlc显示反应完全。反应液浓缩除去溶剂,加入少量饱和碳酸氢钠水溶液,二氯甲烷萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得化合物2c(50mg,粗品),直接用于下一步。
[0240]
步骤(4):(s)-4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((1-甲基吡咯烷-2-基)甲氧基)-1,7-萘啶-3-甲腈(式2化合物)
[0241]
化合物2c(50mg,粗品)溶于二氯甲烷(2.5ml)中,加入水(2.5ml)和碳酸氢钠(78mg,0.93mmol),室温下滴加丙烯酰氯(16mg,0.18mmol),室温反应30分钟,tlc显示反应完全。反应液分液,有机相水洗,无水硫酸钠干燥,浓缩得粗品40mg,prep-hplc纯化得化合物2(10mg,两步收率15%)。
[0242]
lc-ms:m/z=591.2[m h]
。
[0243]1h nmr(400mhz,dmso-d6)δ13.60(s,1h),9.98(s,1h),7.93(d,j=6.0hz,1h),7.78(s,1h),7.65-7.59(m,2h),6.91(dd,j=16.8,10.4hz,1h),6.20(dd,j=16.4,2.0hz,1h),5.77(dd,j=10.4,2.0hz,1h),4.89(dd,j=12.0,2.4hz,1h),4.61(dd,j=12.4,4.0hz,1h),3.96-3.82(m,5h),3.75(s,4h),3.68-3.60(m,1h),3.20-3.12(m,1h),3.05(d,j=4.0hz,3h),2.35-2.25(m,1h),2.11-1.91(m,3h),(92.60%purity by hplc)。
[0244]
实施例4
[0245]
4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2-乙基-1,2,3,4-四氢异喹啉-5-基)氧基)-1,7-萘啶-3-甲腈(式3化合物)
[0246]
[0247]
制备方法:
[0248][0249]
步骤(1):2-乙基-5-羟基异喹啉碘盐3a
[0250]
5-羟基异喹啉1a(2.6g,17.91mmol)溶于乙醇(30ml)中,滴加碘乙烷(5.0ml,62.5mmol),升温至50℃反应30小时,tlc显示基本反应完全。反应液浓缩得固体,加入乙醇溶解后再次浓缩得化合物3a(3.81g,粗品),直接用于下一步。
[0251]
步骤(2):2-乙基-1,2,3,4-四氢异喹啉-5-醇3b
[0252]
化合物3a(3.81g,粗品)溶于乙醇(30ml)中,冷却至0℃,分批加入硼氢化钠(3.28g,86.70mmol),自然升至室温反应过夜,tlc显示反应完全。反应液加水淬灭,二氯甲烷萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得化合物3b(2.93g,两步收率92.4%)。
[0253]
lc-ms:m/z=178.2[m h]
。
[0254]
步骤(3):4-(8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-3-氰基-2-((2-乙基-1,2,3,4-四氢异喹啉-5-基)氧基)-1,7-萘啶-4-基)哌嗪-1-甲酸叔丁酯3c
[0255]
化合物3b(57mg,0.322mmol)溶于n,n-二甲基甲酰胺(15ml)中,加入化合物1f(125mg,0.162mmol)和碳酸铯(281mg,0.862mmol),室温反应1小时,tlc显示反应完全。反应液加水稀释,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得化合物3c(150mg,粗品),直接用于下一步。
[0256]
lc-ms:m/z=699.3[m h]
[0257]
步骤(4):8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2-乙基-1,2,3,4-四氢异喹啉-5-基)氧基)-4-(哌嗪-1-基)-1,7-萘啶-3-甲腈3d
[0258]
化合物3c(150mg,粗品)溶于二氯甲烷(10ml)中,室温滴加三氟乙酸(10ml),加完室温反应1小时,tlc显示反应完全。反应液加入二氯甲烷稀释,饱和碳酸钠水溶液调节至中性,二氯甲烷萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得得化合物3d(102mg,粗品),直接用于下一步。
[0259]
lc-ms:m/z=599.3[m h]
。
[0260]
步骤(5):4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2-乙基-1,2,3,4-四氢异喹啉-5-基)氧基)-1,7-萘啶-3-甲腈(式3化合物)
[0261]
化合物3d(102mg,粗品)溶于二氯甲烷(10ml)中,加入水(10ml)和碳酸氢钠(76mg,0.905mmol),滴加丙烯酰氯(15mg,0.16mmol)的二氯甲烷(2ml)溶液,室温反应20分钟,lc-ms显示反应完全。反应液加水稀释,二氯甲烷萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品prep-hplc纯化得化合物3(15mg,三步收率14.2%)。
[0262]
lc-ms:m/z=653.3[m h]
。
[0263]1h nmr(400mhz,dmso-d6)δ13.49(s,1h),7.92(d,j=6.0hz,1h),7.72-7.60(m,2h),7.51(d,j=8.8hz,1h),7.18(t,j=8.0hz,1h),7.11(d,j=7.6hz,1h),6.98-6.90(m,2h),6.20(dd,j=2.4,16.4hz,1h),5.77(dd,j=2.4,10.0hz,1h),3.91-3.80(m,8h),3.57(s,2h),2.63(s,4h),2.50-2.46(m,2h),1.07-1.04(t,j=7.2hz,3h),(97.85%purity by hplc)。
[0264]
实施例5
[0265]
4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2-甲基-1,2,3,4-四氢异喹啉-8-基)氧基)-1,7-萘啶-3-甲腈(式4化合物)
[0266][0267]
制备方法:
[0268][0269]
步骤(1):2-甲基-5-羟基异喹啉碘盐4a
[0270]
5-羟基异喹啉1a(1.02g,7.03mmol)溶于甲醇(20ml)中,滴加碘甲烷(2.0ml,32.1mmol),升温至50℃反应30小时,tlc显示基本反应完全。反应液浓缩得固体,加入甲醇溶解后再次浓缩得化合物4a(1.23g,粗品),直接用于下一步。
[0271]
步骤(2):2-甲基-1,2,3,4-四氢异喹啉-8-醇4b
[0272]
化合物4a(1.23g,粗品)溶于甲醇(30ml)中,冷却至0℃,分批加入硼氢化钠(1.53g,40.4mmol),自然升至室温反应过夜,tlc显示反应完全。反应液加水淬灭,二氯甲烷萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得化合物4b(722mg,两步收率63%)。
[0273]
lc-ms:m/z=164.2[m h]
。
[0274]
步骤(3):4-(8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-3-氰基-2-((2-甲基-1,2,3,4-四氢异喹啉-8-基)氧基)-1,7-萘啶-4-基)哌嗪-1-甲酸叔丁酯4c
[0275]
化合物4b(63mg,0.386mmol)溶于n,n-二甲基甲酰胺(15ml)中,加入化合物1f(136mg,0.176mmol)和碳酸铯(281mg,0.862mmol),室温反应1小时,tlc显示反应完全。反应液加水稀释,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得化合物4c(150mg,粗品),直接用于下一步。
[0276]
步骤(4):8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2-甲基-1,2,3,4-四氢异喹啉-8-基)氧基)-4-(哌嗪-1-基)-1,7-萘啶-3-甲腈4d
[0277]
化合物4c(150mg,粗品)溶于二氯甲烷(10ml)中,室温滴加三氟乙酸(10ml),加完室温反应1小时,tlc显示反应完全。反应液加入二氯甲烷稀释,饱和碳酸钠水溶液调节至中性,二氯甲烷萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得得化合物4d(104mg,粗品),直接用于下一步。
[0278]
步骤(5):4-(4-甲基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2-甲基-1,2,3,4-四氢异喹啉-8-基)氧基)-1,7-萘啶-3-甲腈(式4化合物)
[0279]
化合物4d(102mg,粗品)溶于二氯甲烷(10ml)中,加入水(10ml)和碳酸氢钠(76mg,0.905mmol),滴加丙烯酰氯(15mg,0.16mmol)的二氯甲烷(2ml)溶液,室温反应20分钟,lc-ms显示反应完全。反应液加水稀释,二氯甲烷萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品prep-hplc纯化得化合物4(11.7mg,三步收率10.4%)。
[0280]
lc-ms:m/z=639.2[m h]
。
[0281]1h nmr(400mhz,dmso-d6)δ13.49(s,1h),7.93(d,j=6.0hz,1h),7.67(d,j=6.0hz,1h),7.51(d,j=8.8hz,1h),7.63(s,1h),7.20(t,j=8.0hz,1h),7.12(d,j=8.0hz,1h),7.04(d,j=7.2hz,1h),6.96-6.89(m,1h),6.21(dd,j=2.0,16.4hz,1h),5.77(dd,j=2.0,10.8hz,1h),3.92-3.81(m,8h),3.58-3.49(m,2h),2.95-2.84(m,2h),2.69(br,2h),2.38(s,3h),(91.32%purity by hplc)。
[0282]
实施例6
[0283]
4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-(((3r,4r)-4-甲氧基-1-甲基吡咯烷-3-基)氧基)-1,7-萘啶-3-甲腈(式5-1化合物)和4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-(((3s,4s)-4-甲氧基-1-甲基吡咯烷-3-基)氧基)-1,7-萘啶-3-甲腈(式5-2化合物)
[0284][0285]
制备方法:
[0286][0287]
步骤(1):3-羟基-4-甲氧基吡咯烷-1-甲酸叔丁酯5b
[0288]
6-氧杂-3-氮杂二环[3.1.0]己烷-3-甲酸叔丁酯5a(10.6g,57.2mmol)溶于甲醇(50ml)中,氮气保护下,加入甲醇钠(3.71g,68.7mmol),升温至60℃反应5小时,tlc监测原料反应完全。反应液冷却至室温,加水稀释,乙酸乙酯萃取,合并有机相,水洗,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得白色固体化合物5b(8.0g,收率65%)。
[0289]
步骤(2):4-甲氧基-1-甲基吡咯烷-3-醇5c
[0290]
将氢化铝锂(665mg,17.5mmol)分散至干燥的四氢呋喃(30ml)中,降温至0℃,氮气保护下,滴加化合物5b(1.9g,8.75mmol)的四氢呋喃(15ml)溶液,滴加完毕,升温至50℃加热反应2小时,tlc监测原料反应完全。反应液冷却至室温,依次加入水(0.65ml)、15%氢氧化钠溶液(0.65ml)和水(1.95ml)淬灭,过滤,滤液浓缩得黄色油状物化合物5c(1.1g,收率99%)。
[0291]
步骤(3):4-((4-(4-(叔丁氧基羰基)哌嗪-1-基)-3-氰基-2-((4-甲氧基-1-甲基吡咯烷-3-基)氧基)-1,7-萘啶-8-基)氧基)-5-氯-6-氟-1h-吲唑-1-甲酸叔丁酯5d
[0292]
化合物1f(800mg,1.04mmol)溶于n,n-二甲基甲酰胺(30ml)中,依次加入化合物5c(204mg,1.56mmol)的n,n-二甲基甲酰胺(10ml)溶液和碳酸钾(728mg,5.27mmol),室温搅拌5小时,tlc监测原料消失。反应液加水稀释,乙酸乙酯萃取,合并有机相,水洗,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得化合物5d(435mg,收率56%)。
[0293]
lc-ms:m/z=753.3[m h]
[0294]
步骤(4):4-(8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-3-氰基-2-((4-甲氧基-1-甲基吡咯烷-3-基)氧基)-1,7-萘啶-4-基)哌嗪-1-甲酸叔丁酯5e
[0295]
化合物5d(435mg,0.58mmol)溶于三氟乙酸(10ml)和二氯甲烷(5ml)的混合溶液中,室温搅拌0.5小时,tlc监测原料消失。反应液浓缩,依次加入水(5ml)和饱和碳酸氢钠溶液(5ml),二氯甲烷萃取,合并有机相,水洗,饱和食盐水洗,无水硫酸钠干燥,浓缩得化合物5e(300mg,粗品),直接用于下一步。
[0296]
lc-ms:m/z=553.3[m h]
。
[0297]
步骤(5):4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-(((3r,4r)-4-甲氧基-1-甲基吡咯烷-3-基)氧基)-1,7-萘啶-3-甲腈(式5-1化合物)&4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-(((3s,4s)-4-甲氧基-1-甲基吡咯烷-3-基)氧基)-1,7-萘啶-3-甲腈(式5-2化合物)
[0298]
化合物5e(300mg,粗品)溶于二氯甲烷(10ml)中,依次加入水(10ml)和碳酸氢钠(90mg,1.07mmol),冷却至0℃,缓慢滴加丙烯酰氯(24mg,0.27mmol)的二氯甲烷(2ml)溶液,室温搅拌5分钟,tlc监测反应完全。反应液加水稀释,分液,二氯甲烷萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得化合物5(50mg,两步收率14%))。化合物5手性拆分(赛璐ad-h,21.2*250mm,5um,20ml/min,ipa:hexane=20:80)得化合物5-1(1号峰,rt 28.7min)(12mg,收率48%)和化合物5-2(2号峰,rt 32.9min)(10mg,收率40%)。
[0299]
化合物5-1
[0300]
lc-ms:m/z=607.2[m h]
。
[0301]1h nmr(400mhz,dmso-d6)δ13.61(s,1h),10.79(s,1h),7.93(d,j=5.6hz,1h),7.79(s,1h),7.65(d,j=6.0hz,1h),7.60(d,j=8.8hz,1h),6.91(dd,j=16.8,10.4hz,
1h),6.20(dd,j=16.4,2.0hz,1h),5.84(d,j=4.4hz,1h),5.76(dd,j=10.4,2.0hz,1h),4.37(s,1h),3.95-3.70(m,9h),3.42(s,6h),2.97(s,3h),(90.93%purity by hplc)。
[0302]
化合物5-2
[0303]
lc-ms:m/z=607.2[m h]
。
[0304]1h nmr(400mhz,dmso-d6)δ13.61(s,1h),10.77(s,1h),7.93(d,j=5.6hz,1h),7.79(s,1h),7.65(d,j=6.0hz,1h),7.60(d,j=8.8hz,1h),6.91(dd,j=16.8,10.4hz,1h),6.19(dd,j=16.4,2.0hz,1h),5.83(d,j=4.4hz,1h),5.76(dd,j=10.4,2.0hz,1h),4.36(s,1h),3.94-3.70(m,9h),3.40(s,6h),2.96(s,3h),(92.27%purity by hplc)。
[0305]
实施例7
[0306]
4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2,4,4-三甲基-1,2,3,4-四氢异喹啉-5-基)氧基)-1,7-萘啶-3-甲腈(式6化合物)
[0307][0308]
制备方法:
[0309]
[0310]
步骤(1):2-(2-溴苯基)-2-甲基丙腈6b
[0311]
将邻溴氰苄6a(10.0g,51.01mmol)溶于四氢呋喃(100ml)中,降温至0℃,加入氢化钠(6.1g,0.15mol,60%),加完升至室温反应2小时。再次降温至0℃,加入碘甲烷(14.2g,0.10mol),加完升至室温反应2小时,tlc显示反应完全。反应液倒入水中,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得无色液体化合物6b(10.2g,收率89%)。
[0312]1h nmr(400mhz,dmso-d6)δ7.74(d,j=8.0hz,1h),7.56(dd,j=8.0,1.2hz,1h),7.47(t,j=8.0hz,1h),7.33-7.29(m,1h),1.82(s,6h)。
[0313]
步骤(2):2-(2-溴苯基)-2-甲基丙烷-1-胺6c
[0314]
化合物6b(11.5g,51.32mmol)溶于四氢呋喃(100ml)中,降温至0℃,加入二甲硫醚硼烷(15ml,150mmol,10m),加完升温至回流反应18小时。反应液浓缩除去溶剂,加入甲醇(50ml),升温至回流反应12小时,tlc显示反应完成。反应液冷却至室温,倒入水中,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化(二氯甲烷:甲醇=50:1,0.5%氨水)得无色透明液体化合物6c(5.1g,收率44%)。
[0315]
lc-ms:m/z=228.1[m h]
。
[0316]1h nmr(400mhz,cdcl3)δ7.60(d,j=7.6hz,1h),7.56(dd,j=7.6,0.8hz,1h),7.28-7.26(m,1h),7.08-7.04(m,1h),3.22(s,2h),1.48(s,6h)。
[0317]
步骤(3):(2-(2-溴苯基)-2-甲基丙基)氨基甲酸甲酯6d
[0318]
化合物6c(5.1g,22.36mmol)溶于干燥的四氢呋喃(50ml)中,室温下加入三乙胺(3.4g,33.60mmol),降温至0℃,加入氯甲酸甲酯(2.5g,26.46mmol),加完升至室温反应2小时,tlc显示反应完全。反应液倒入水中,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得黄色透明粘稠液体化合物6d(5.1g,收率80%)。
[0319]
lc-ms:m/z=286.1[m h]
。
[0320]
步骤(4):5-溴-4,4-二甲基-3,4-二氢异喹啉-1(2h)-酮6e
[0321]
化合物6d(5.1g,17.82mmol)溶于多聚磷酸(30ml)中,升温至140℃反应2小时,tlc显示反应完全。反应液冷却至室温,倒入水中,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得黄色固体化合物6e(1.5g,收率33%)。
[0322]1h nmr(400mhz,dmso-d6)δ8.18(br,1h),7.95(d,j=7.6hz,1h),7.77(d,j=7.6hz,1h),7.27(t,j=7.6hz,1h),3.15(d,j=2.8hz,2h),1.46(s,6h)。
[0323]
步骤(5):5-溴-2,4,4-三甲基-3,4-二氢异喹啉-1(2h)-酮6f
[0324]
化合物6e(1.0g,3.93mmol)溶于四氢呋喃中,降温至0℃,加入氢化钠(250mg,6.25mmol,60%),加完升至室温反应0.5小时,再次降温至0℃,加入碘甲烷(682mg,4.80mmol),加完升至室温反应1小时,tlc显示反应完全。反应液倒入水中,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得黄色固体化合物6f(1.1g,粗品),直接用于下一步。
[0325]
lc-ms:m/z=268.1[m h]
。
[0326]
步骤(6):5-羟基-2,4,4-三甲基-3,4-二氢异喹啉-1(2h)-酮6g
[0327]
化合物6f(1.1g,粗品)溶于1,4-二氧六环(8ml)和水(2ml)的混合溶剂中,室温下加入氢氧化钾(690mg,12.30mmol),pd2(dba)3(37mg,0.04mmol)和t-buxphos(17mg,
0.04mmol),氮气保护下,升温至90℃反应2小时,tlc显示反应完全。反应液冷却至室温,倒入水中,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得白色固体化合物6g(400mg,两步收率50%)。
[0328]
lc-ms:m/z=206.1[m h]
。
[0329]
步骤(7):2,4,4-三甲基-1,2,3,4-四氢异喹啉-5-醇6h
[0330]
化合物6g(400mg,1.95mmol)溶于四氢呋喃中,降温至0℃,加入四氢铝锂(230mg,6.06mmol),加完升至室温反应0.5小时,tlc显示反应完成。反应液加水(0.3ml),室温搅拌0.5小时,过滤,滤液浓缩得白色固体化合物6h(300mg,粗品),直接用于下一步。
[0331]
lc-ms:m/z=192.2[m h]
。
[0332]
步骤(8):4-((4-(4-(叔丁氧基羰基)哌嗪-1-基)-3-氰基-2-((2,4,4-三甲基-1,2,3,4-四氢异喹啉-5-基)氧基)-1,7-萘啶-8-基)氧基)-5-氯-6-氟-1h-吲唑-1-甲酸叔丁酯6i
[0333]
化合物6h(46mg,粗品)溶于n,n-二甲基甲酰胺(5ml)中,室温下加入化合物1f(150mg,0.19mmol)和碳酸铯(126mg,0.39mmol),升温至35℃反应0.5小时,tlc显示反应完全。反应液倒入水中,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得白色固体化合物6i(80mg,收率51%)。
[0334]
lc-ms:m/z=713.3[m-boc h]
。
[0335]
步骤(9):8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-4-(哌嗪-1-基)-2-((2,4,4-三甲基-1,2,3,4-四氢异喹啉-5-基)氧基)-1,7-萘啶-3-甲腈6j
[0336]
化合物6i(80mg,0.10mmol)溶于二氯甲烷(4ml)中,加入三氟乙酸(2ml),室温反应2小时,tlc显示反应完全。反应液浓缩,饱和碳酸氢钠溶液调节ph=8-9,乙酸乙酯萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得黄色固体化合物6j(68mg,粗品),直接用于下一步。
[0337]
步骤(10):4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-((2,4,4-三甲基-1,2,3,4-四氢异喹啉-5-基)氧基)-1,7-萘啶-3-甲腈(式6化合物)
[0338]
化合物6j(68mg,粗品)溶于二氯甲烷(5ml)中,依次加入水(5ml),碳酸氢钠(90mg,1.07mmol)和丙烯酰氯(12mg,0.13mmol),室温反应0.5小时,lcms显示反应完全。反应液二氯甲烷萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品prep-hplc纯化得淡黄色固体化合物6(35mg,两步收率46%)。
[0339]
lc-ms:m/z=667.2[m h]
。
[0340]1h nmr(400mhz,dmso-d6)δ13.53(br,1h),9.87(s,1h),7.96(d,j=5.6hz,1h),7.68(d,j=6.0hz,1h),7.62(s,1h),7.53(d,j=8.8hz,1h),7.34-7.28(m,2h),7.09(dd,j=6.8,2.0hz,1h),6.89-6.96(m,1h),6.20(dd,j=16.4,2.0hz,1h),5.77(dd,j=10.4,2.0hz,1h),4.49-4.52(m,1h),4.33-4.39(m,1h),3.83-3.92(m,9h),3.23-3.29(m,1h),2.98(s,3h),1.56(s,3h),1.31(s,3h),(90.80%purity by hplc)。
[0341]
实施例8
[0342]
4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-(((3s,4r)-4-二氟甲基-1-甲基吡咯烷-3-基)氧基)-1,7-萘啶-3甲腈(式7-1化合物)和4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-(((3r,4s)-4-二氟甲基-1-甲基吡咯
烷-3-基)氧基)-1,7-萘啶-3甲腈(式7-2化合物)
[0343][0344]
制备方法:
[0345][0346]
步骤(1):(3r,4s)-4-(二氟甲基)-1-甲基吡咯烷-3-基4-硝基苯甲酸酯7b
[0347]
(3s,4s)-4-(二氟甲基)-1-甲基吡咯烷-3-醇7a(325mg,2.15mmol),间硝基苯甲酸(719mg,4.30mmol)和三苯基膦(1.1g,4.19mmol)置于三口瓶中,氮气保护下,加入干燥四氢呋喃(15ml),冷却至0℃,加入偶氮二甲酸二异丙酯(870mg,4.30mmol),加完升至室温反应过夜,tlc监控反应完全。反应液加水,二氯甲烷萃取,合并有机相,水洗、饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱层析纯化得化合物7b(500mg,收率77%)。
[0348]
lc-ms:m/z=301.1[m h]
。
[0349]
步骤(2):(3r,4r)-4-(二氟甲基)-1-甲基吡咯烷-3-醇7c
[0350]
化合物7b(500mg,1.66mmol)溶于甲醇(10ml)中,加入碳酸钾(460mg,3.33mmol),室温搅拌过夜,tlc显示反应完全。反应液浓缩去除溶剂,加入正己烷(20ml),搅拌10分钟,过滤,滤饼正己烷洗涤,滤液浓缩得化合物7c(130mg,粗品),直接用于下一步。
[0351]
lc-ms:m/z=152.1[m h]
。
[0352]
步骤(3):4-((4-(4-(叔丁氧基羰基)哌嗪-1-基)-3-氰基-2-(((3s,4r)-4-(二氟甲基)-1-甲基吡咯烷-3-基)氧基)-1,7-萘啶-8-基)氧基)-5-氯-6-氟-1h-吲唑-1-甲叔丁酯7d
[0353]
化合物1e(400mg,0.62mmol),化合物7c(130mg,粗品)和三苯基膦(328mg,1.25mmol)置于三口瓶中,氮气保护下,加入干燥四氢呋喃(14ml),冷却至0℃,加入偶氮二甲酸二异丙酯(253mg,1.25mmol),加完室温反应过夜,tlc监控反应完全。反应液加水,二氯甲烷萃取,合并有机相,水洗、饱和食盐水洗,无水硫酸钠干燥,浓缩,粗品硅胶柱(2%ma in二氯甲烷)纯化得化合物7d(180mg,收率37%)。
[0354]
lc-ms:m/z=773.3[m h]
。
[0355]
步骤(4):8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-(((3s,4r)-4-(二氟甲基)-1-甲基吡咯烷-3-基)氧基)-4-(哌嗪-1-基)-1,7-萘啶-3-甲腈7e
[0356]
化合物7d(60mg,0.08mmol)溶于二氯甲烷(1ml)中,加入三氟乙酸(1ml),室温反应1小时,tlc显示反应完全。反应液浓缩除去溶剂,残留固体加入饱和碳酸氢钠水溶液(2ml),二氯甲烷萃取,合并有机相,饱和食盐水洗,无水硫酸钠干燥,浓缩得标题化合物7e(40mg,粗品),直接用于下一步。
[0357]
lc-ms:m/z=573.2[m h]
。
[0358]
步骤(5):4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-(((3s,4r)-4-二氟甲基-1-甲基吡咯烷-3-基)氧基)-1,7-萘啶-3甲腈(式7-1化合物)&4-(4-丙烯酰基哌嗪-1-基)-8-((5-氯-6-氟-1h-吲唑-4-基)氧基)-2-(((3r,4s)-4-二氟甲基-1-甲基吡咯烷-3-基)氧基)-1,7-萘啶-3甲腈(式7-2化合物)
[0359]
化合物7e(40mg,粗品)溶于二氯甲烷(2.5ml)中,加入水(2.5ml)和碳酸氢钠(59mg,0.70mmol),室温下滴加丙烯酰氯(10mg,0.09mmol),室温反应30分钟,tlc显示反应完全。反应液分液,有机相水洗,无水硫酸钠干燥,浓缩得粗品40mg,prep-hplc纯化得标题化合物7(20mg,两步收率41%)。化合物构型及性质需进一步检测。化合物7手性拆分(大赛璐
[0360]
ad-h,20*250mm,5um,20ml/min,etoh:hexane=20:80)得化合物7-1(1号峰,rt22.7min)(2.0mg,收率10%)和化合物7-2(2号峰,rt 29.1min)(2.0mg,收率10%)。
[0361]
化合物7-1:
[0362]
lc-ms:m/z=627.2[m h]
。
[0363]1h nmr(400mhz,cd3od)δ7.87(d,j=6.0hz,1h),7.71(s,1h),7.64(d,j=6.0hz,1h),7.41(d,j=8.4hz,1h),6.86(dd,j=16.8,10.8hz,1h),6.29(dd,j=16.8,2.0hz,1h),5.82(dd,j=10.8,1.6hz,1h),5.53-5.45(m,1h),4.05-3.97(m,4h),3.82(s,4h),3.25-3.19(m,1h),3.19-3.10(m,1h),2.81(dd,j=11.2,6.4hz,1h),2.62-2.54(m,1h),2.44(s,3h),2.19(t,j=7.2hz,1h),2.05-2.00(m,1h),(90.50%purity by hplc)。
[0364]
化合物7-2:
[0365]
lc-ms:m/z=627.2[m h]
。
[0366]1h nmr(400mhz,cd3od)δ7.87(d,j=6.0hz,1h),7.71(s,1h),7.63(d,j=6.0hz,1h),7.40(dd,j=8.8,1.2hz,1h),6.89-6.81(m,1h),6.28(dd,j=16.8,2.0hz,1h),5.82(dd,j=10.4,1.6hz,1h),5.56-5.45(m,1h),4.03-3.90(m,4h),3.82(s,4h),3.27-3.22(m,1h),3.19-3.13(m,1h),2.84(dd,j=11.6,6.4hz,1h),2.66-2.57(m,1h),2.46(s,3h),2.18(t,j=7.2hz,1h),2.07-1.98(m,1h),(88.16%purity by hplc)。
[0367]
实验例1.化合物对miapaca-2细胞增殖抑制的ic
50
测定
[0368]
本发明中使用的人胰腺癌miapaca-2细胞(crl-1420)购自american type culture collection(atcc)。细胞在2.5%马血清、10%胎牛血清及1%双抗的dmem培养基,于37℃,5%co2的环境中生长。
[0369]
本发明的实施例2-8制备的化合物对体外培养的miapaca-2细胞增殖抑制作用通过以下方法进行测定:
[0370]
1)细胞接种:取对数生长期状态良好的胰腺癌miapaca-2细胞重悬于完全培养基,以3000个/孔接种到96孔板中,每孔180μl,在37℃、5%co2条件下培养过夜。
[0371]
2)加药:将需要测试的化合物以完全培养基进行梯度稀释,取20μl稀释的化合物加入到180μl的细胞中,使化合物终浓度为10000、3000、1000、300、100、30、10、3、1nm,同时设相应的溶媒对照。置于37℃、5%co2细胞培养箱中培养120小时。
[0372]
3)检测:以10%三氯乙酸固定细胞,去上清洗涤后,每孔加入100μl 4mg/ml的srb溶液染色15分钟(sigma,s1402-25g),最后加入加150μl 10mmtris溶液溶解srb,tecan spark多功能酶标仪读取od510值。
[0373]
4)计算:以下列公式计算细胞生长抑制率:抑制率=(od值对照孔-od值给药孔)/od值对照孔
×
100%。用graphpad prism 5.0软件根据化合物浓度与对应的抑制率计算ic
50
值。检测结果详见下表2。
[0374]
表2本发明所述化合物对miapaca-2细胞增殖抑制的ic50(nm)
[0375]
化合物编号miapaca-2化合物编号miapaca-2mrtx-8491.95-11.3119.65-211.7294.0645.6334.17-130.74114.97-2202
[0376]
其中,mrtx-849为对照品,购自美国mirati公司。
[0377]
表2表明,本发明所述的kras突变体g12c抑制剂具有较好的抑制miapaca-2细胞的活性,其中化合物5-1与mrtx-849活性相当。
[0378]
实验例2.化合物对kras下游信号分子erk1/2磷酸化水平的影响
[0379]
本发明的实施例2-8制备的化合物对miapaca-2细胞中erk1/2磷酸化水平的影响通过以下方法检测:
[0380]
1)细胞接种:取对数生长期状态良好的miapaca-2细胞以5*105个/孔接种到六孔板中,在37℃、5%co2条件下培养过夜。
[0381]
2)加药:将需要测试的化合物以完全培养基进行梯度稀释后,加入细胞中,使化合物的终浓度为1000、100、10、1nm。置于37℃、5%co2细胞培养箱中培养24小时。
[0382]
3)蛋白样品制备:0.25%胰酶消化后,收集细胞悬液,500g离心5分钟,弃上清,pbs洗涤3次,以1
×
sds凝胶上样缓冲液(50mm tris-hcl(ph 6.8),100mm dtt,2%sds,10%甘油,0.1%溴酚蓝)100μl裂解细胞。细胞裂解物在100℃中加热10分钟变性。
[0383]
4)western blot:将蛋白样品进行sds-page电泳,电泳结束后,用湿转系统将蛋白转移至pvdf膜,将pvdf膜置于封闭液(5%脱脂奶粉稀释于tbs/t)中室温封闭1小时,然后i,ii抗反应;洗膜后,用immobilon western hrp substrate luminal reagent试剂发色,
western blot成像仪(tanon,4600)拍照。其中,所用的抗体信息为:p-erk1/2(cst:4370);β-tubulin(cst:2146)。
[0384]
本发明制备的化合物对miapaca-2细胞中erk1/2磷酸化水平影响的结果见图1。由图1可知,本发明制备的化合物对miapaca-2细胞中erk1/2的磷酸化具有明显抑制作用,抑制活性呈浓度梯度依赖,活性与mrtx849大致相当。
[0385]
申请人声明,本发明通过上述实施例来说明本发明的一类作为kras突变体g12c抑制剂的萘啶甲腈类衍生物、其制备方法及其应用,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
[0386]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。