1.本技术属于有机化学合成技术领域,特别是涉及一种烷基取代芴及衍生物及其制备方法。
背景技术:
2.芴,是一种有机物,用于制医药(制造抗痉挛药、镇静药,镇痛药,降血压药)、染料(代替蒽醌合成阴丹士林染料);合成杀虫剂、除草剂;制抗冲击有机玻璃和芴醛树脂;用作湿润剂、洗涤剂、液体闪光剂、消毒剂等。用作有机合成原料;可制成三硝基芴酮,用于静电复印;合成芳基透明尼龙。
3.芴及其衍生物由于本身特殊的结构,使得其具有多方面的应用,如合成药物中应用,有机电至发光材料中的应用,高分子材料,有机柔性材料领域等,在其机构的9位引入不同的活性基团,会在不同的领域都有不同的应用和更优异的性能,由于近年来科学家们对有机电至发光材料的不断研究和开发,9位引入各种取代基需要越来越高的纯度和收率,因此需要对工艺不断的进行优化和改进。
技术实现要素:
4.1.要解决的技术问题
5.基于现有的烷基取代芴及其衍生物反应复杂,产率低,后处理复杂的问题,本技术提供了一种烷基取代芴及其衍生物的制备方法及其衍生物。
6.2.技术方案
7.为了达到上述的目的,本技术提供了一种烷基取代芴及其衍生物的制备方法,以2-溴联苯或者2-溴联苯衍生物为原料在锂试剂存在条件下发生反应,再加入无α氢取代烷基酮发生取代反应后,在酸性条件下制备得到烷基取代芴或者烷基取代芴衍生物。
8.本技术提供的另一种实施方式为:通惰性气体下,在反应容器中加入溶剂,然后加入2-溴联苯或者2-溴联苯衍生物,降温至-50℃~-90℃,加入无机锂试剂,反应0.5~1小时后加入无α氢取代烷基酮,反应1~3小时后升温至-50℃~0℃,加酸性试剂淬灭,石油醚煮洗后得到高纯度的中间体s;向反应器中加入s,第二溶剂和脱水剂,在80℃-100℃下反应2~5小时,降温,抽滤,得到高纯度产品烷基取代芴或者烷基取代芴衍生物。
9.本技术提供的另一种实施方式为:所述溶剂为乙醚,2-甲基四氢呋喃或者四氢呋喃;所述无α氢取代烷基酮包括2,2,4,4-四甲基-3-戊酮,二(1,2-环己烯基)甲酮或者二(1,2-环戊烯)甲酮。
10.本技术提供的另一种实施方式为:所述无机锂试剂为正丁基锂,仲丁基锂,6-甲基硅基氨基锂或者二异丙基氨基锂。
11.本技术提供的另一种实施方式为:所述溶剂为四氢呋喃,所述四氢呋喃当量为2-溴联苯或者2-溴联苯衍生物5~15当量;所述无机碱为仲丁基锂,所述仲丁基锂当量为2-溴联苯或者2-溴联苯衍生物1~2当量。
12.本技术提供的另一种实施方式为:所述酸性试剂为醋酸,磷酸,盐酸或者对甲苯磺酸。
13.本技术提供的另一种实施方式为:所述第二溶剂为冰醋酸,n,n-二甲基甲酰胺,乙醇或者磷酸;优选为冰醋酸,所述冰醋酸当量为中间体s的5~10当量。
14.本技术提供的另一种实施方式为:所述脱水剂为盐酸,多聚磷酸,亚磷酸钠或者硫酸,优选为盐酸。
15.本技术还提供一种烷基取代芴及其衍生物,采用所述的烷基取代芴及其衍生物的制备方法制得,所述烷基取代芴及其衍生物通式为:
[0016][0017]
其中,r1为无α氢取代烷基酮,r2为无α氢取代烷基酮,所述r3为氢,氯或氟;所述无α氢取代烷基酮中烷基为c4~c40,优选为c4~c12。
[0018]
本技术还提供一种烷基取代芴及其衍生物的应用,将所述的烷基取代芴及其衍生物应用合成药物,有机电至发光材料,高分子材料或者有机柔性材料。
[0019]
3.有益效果
[0020]
与现有技术相比,本技术提供的烷基取代芴及其衍生物的制备方法及其衍生物的有益效果在于:
[0021]
本技术提供的烷基取代芴及其衍生物的制备方法,为一种反应简单易操作,产率高,后处理简单,废水少,更环保的制备烷基取代芴及其衍生物的合成方法。
附图说明
[0022]
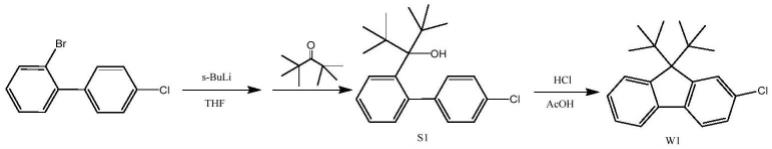
图1为本技术的烷基取代芴及其衍生物合成路线示意图。
具体实施方式
[0023]
在下文中,将参考附图对本技术的具体实施例进行详细地描述,依照这些详细的描述,所属领域技术人员能够清楚地理解本技术,并能够实施本技术。在不违背本技术原理的情况下,各个不同的实施例中的特征可以进行组合以获得新的实施方式,或者替代某些实施例中的某些特征,获得其它优选的实施实施方式。
[0024]
参见图1,本技术提供一种烷基取代芴及其衍生物的制备方法,以2-溴联苯或者2-溴联苯衍生物为原料在锂试剂存在条件下发生反应,再加入无α氢取代烷基酮发生取代反应后,在酸性条件下制备得到烷基取代芴或者烷基取代芴衍生物。
[0025]
以2-溴联苯或者2-溴联苯衍生物为原料在锂试剂存在的条件下进行锂卤交换,得到芳基锂试剂,再与体系中的烷基酮发生亲核取代反应,得到中间体s,中间体s在酸性条件下脱水关环制备得到烷基取代芴或者烷基取代芴衍生物。
[0026]
进一步地,通惰性气体下,在反应容器中加入溶剂,然后加入2-溴联苯或者2-溴联苯衍生物,降温至-50℃~-90℃,加入无机锂试剂,反应0.5~1小时后加入无α氢取代烷基酮,反应1~3小时后升温至-50℃~0℃,加酸性试剂淬灭,石油醚煮洗后得到高纯度的中间
体s;向反应器中加入s,第二溶剂和脱水剂,在80℃-100℃下反应2~5小时,降温,抽滤,得到高纯度产品烷基取代芴或者烷基取代芴衍生物。
[0027]
进一步地,所述溶剂为乙醚,2-甲基四氢呋喃或者四氢呋喃。
[0028]
进一步地,所述无机锂试剂为正丁基锂,仲丁基锂,6-甲基硅基氨基锂或者二异丙基氨基锂。
[0029]
进一步地,所述溶剂为四氢呋喃,所述四氢呋喃当量为2-溴联苯或者2-溴联苯衍生物5~15当量;所述无机碱为仲丁基锂,所述仲丁基锂当量为2-溴联苯或者2-溴联苯衍生物1~2当量。
[0030]
进一步地,所述酸性试剂为醋酸,磷酸,盐酸或者对甲苯磺酸。
[0031]
进一步地,所述第二溶剂为冰醋酸,n,n-二甲基甲酰胺,乙醇或者磷酸;优选为冰醋酸,所述冰醋酸当量为中间体s的5~10当量。
[0032]
进一步地,所述脱水剂为盐酸,多聚磷酸,亚磷酸钠或者硫酸,优选为盐酸。
[0033]
本技术还体用一种烷基取代芴及其衍生物,采用所述的烷基取代芴及其衍生物的制备方法制得,所述烷基取代芴及其衍生物通式为:
[0034][0035]
其中,r1为无α氢取代烷基酮,r2为无α氢取代烷基酮,所述r3为氢,氯或氟;所述无α氢取代烷基酮中烷基为c4~c40,优选为c4~c12。
[0036]
本技术还提供一种烷基取代芴及其衍生物的应用,将所述的烷基取代芴及其衍生物应用合成药物,有机电至发光材料,高分子材料或者有机柔性材料。
[0037]
本技术所述的制备方法,各步骤的反应条件(如反应温度,溶剂的选择,产物的分离等)可采用本领域的常规可用手段,满足能够实现上述反应历程制得即可。
[0038]
(1)向反应器中加入干燥四氢呋喃,然后加入r3取代2-溴联苯,降温至-70℃,滴加仲丁基锂,反应0.5小时后加入无α氢取代烷基酮,反应1小时后升温至-40℃,加稀盐酸淬灭,抽滤,石油醚煮洗后得到高纯度的中间体s。
[0039]
(2)向反应器中加入s,冰醋酸,盐酸,在80℃下反应4小时,降温,抽滤,得到高纯度产品取代烷基芴及其衍生物。
[0040]
以下实施例用于说明本发明,但不用来限制本发明的范围。本发明中所使用的基本原材料均为常规市售产品。
[0041]
实施例1:
[0042][0043]
(1)通氮气下,在1l玻璃三口烧瓶中加入400ml干燥的四氢呋喃,然后加入2-溴-4
’‑
氯-1,1
’‑
联苯(40g,0.15mol),降温到-90℃,缓慢滴加仲丁基锂(75ml,0.15mol),保温一个小时,然后加入2,2,4,4-四甲基-3-戊酮(21.3g,0.15mol),反应1小时。
[0044]
后处理:升温至-40℃,加入200ml,1mol/l的稀盐酸淬灭,搅拌一小时后抽滤,固体用1l石油醚洗涤后,得到hplc=97.12%含量的中间体s,质量为33.27g收率为67%。
[0045]
(2)向1l玻璃釜中加入33.27g/s,330ml/冰醋酸,80ml/盐酸,加热至130℃反应1小时。
[0046]
后处理:降温至30℃,用抽滤漏斗在负压情况下抽滤,得到粗品,再用100g碳酸钠水溶液洗涤一次,然后用200ml正己烷洗涤一次,得到产品9.8g hplc=98.02%,收率53%。
[0047]
实施例2:
[0048][0049]
(1)通氮气下,在1l玻璃三口烧瓶中加入400ml干燥的2-甲基四氢呋喃,然后加入2-溴-3
’‑
氟-1,1
’‑
联苯(40g,0.16mol),降温到-60℃,缓慢滴加正丁基锂(95.6ml,0.24mol),保温一个小时,然后加入二(1,2-环戊烯)甲酮(30.98g,0.19mol),反应1小时后。
[0050]
后处理:升温至-40℃,加入200ml,1mol/l的稀盐酸淬灭,搅拌一小时后抽滤,固体用1l石油醚洗涤后,得到hplc=96%含量的中间体s,质量为29.27g收率为55%。
[0051]
(2)向1l玻璃釜中加入29.27g/s,250ml/磷酸,40ml/多聚磷酸,加热至130℃反应1小时。
[0052]
后处理:降温至30℃,用抽滤漏斗在负压情况下抽滤,得到粗品,再用100g碳酸钠水溶液洗涤一次,然后用200ml正己烷洗涤一次,得到产品19.11g hplc=97.30%,收率69%。
[0053]
实施例3:
[0054][0055]
(1)通氮气下,在1l玻璃三口烧瓶中加入400ml干燥的四氢呋喃,然后加入2-溴-2
’‑
氯-1,1
’‑
联苯(40g,0.15mol),降温到-70℃,缓慢滴加仲丁基锂(298ml,0.30mol),保温一个小时,然后加入二(1,2-环己烯基)甲酮(42.54g,0.22mol),反应1小时后。
[0056]
后处理:升温至-40℃,加入200ml,1mol/l的氯化铵淬灭,搅拌一小时后抽滤,固体用1l石油醚洗涤后,得到hplc=99%含量的中间体s,质量为34.51g收率为61%。
[0057]
(2)向1l玻璃釜中加入34.51g/s,300ml/冰醋酸,80g/对甲苯磺酸,加热至140℃反应1小时。
[0058]
后处理:降温至30℃,用抽滤漏斗在负压情况下抽滤,得到粗品,再用100g碳酸钠水溶液洗涤一次,然后用200ml正己烷洗涤一次,得到产品8.54g hplc=98.01%,收率51%。
[0059]
实施例4:
[0060][0061]
(1)通氮气下,在1l玻璃三口烧瓶中加入400ml干燥的乙醚,然后加入2-溴-3
’‑
氟-1,1
’‑
联苯(40g,0.16mol),降温到-80℃,缓慢滴加正丁基锂(115.2ml,0.29mol),保温一个小时,然后加入2,4-二氟-2,4-二甲基-3-戊酮(43.21g,0.29mol),反应1小时后。
[0062]
后处理:升温至-40℃,加入200ml,1mol/l的稀盐酸淬灭,搅拌一小时后抽滤,固体用1l石油醚洗涤后,得到hplc=97.5%含量的中间体s,质量为26.28g收率为51%。
[0063]
(2)向1l玻璃釜中加入26.28g/s,200ml/冰醋酸,80ml/盐酸,加热至130℃反应1小时。
[0064]
后处理:降温至30℃,用抽滤漏斗在负压情况下抽滤,得到粗品,再用100g碳酸钠水溶液洗涤一次,然后用200ml正己烷洗涤一次,得到产品9.91g hplc=98.40%,收率66%。
[0065]
实施例5:
[0066][0067]
(1)通氮气下,在1l玻璃三口烧瓶中加入400ml干燥的2-甲基四氢呋喃,然后加入2-溴-4
’‑
氯-1,1
’‑
联苯(40g,0.15mol),降温到-70℃,缓慢滴加二异丙基氨基锂(195ml,0.20mol),保温一个小时,然后加入2,4-二环戊基-2,4-二甲基-3-戊酮(60.10g,0.24mol),反应1小时后。
[0068]
后处理:升温至-40℃,加入200ml,1mol/l的稀盐酸淬灭,搅拌一小时后抽滤,固体用1l石油醚洗涤后,得到hplc=96.8%含量的中间体s,质量为32.34g收率为56%。
[0069]
(2)向1l玻璃釜中加入32.34g/s,300ml/无水乙醇,100ml/盐酸,加热至130℃反应1小时。
[0070]
后处理:降温至30℃,用抽滤漏斗在负压情况下抽滤,得到粗品,再用100g碳酸钠水溶液洗涤一次,然后用200ml正己烷洗涤一次,得到产品9.74g hplc=98.00%,收率63%。
[0071]
上述实施例中实施例2为最佳实施例。
[0072]
尽管在上文中参考特定的实施例对本技术进行了描述,但是所属领域技术人员应当理解,在本技术公开的原理和范围内,可以针对本技术公开的配置和细节做出许多修改。本技术的保护范围由所附的权利要求来确定,并且权利要求意在涵盖权利要求中技术特征的等同物文字意义或范围所包含的全部修改。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。