1.本发明属于蛋白质药物技术领域,涉及一种艾塞那肽变体的制备方法,还涉及一种聚乙二醇化艾塞那肽变体的制备方法。
背景技术:
2.随着经济社会的发展、人口平均寿命的延长和生活方式的变化,糖尿病已经成为世界各国主要的卫生保健问题。无论在发达国家或是发展中国家,糖尿病的发病率都在急剧上升。糖尿病是由于胰腺分泌功能缺陷或胰岛素作用缺陷引起的,以血糖升高为特征的代谢性疾病。糖尿病患者若血糖长期控制不佳,可导致器官组织损伤,成为“万病之源”,伴发各种器官,尤其是眼、心、血管、肾、神经损害或器官功能不全或衰竭,严重可致患者残疾或者死亡。根据发病机制不同,糖尿病主要分为i型糖尿病、ii型糖尿病、妊娠期糖尿病和其他类型糖尿病。ii型糖尿病患者是糖尿病的主体,占比约为90%。ii型糖尿病的特点是胰岛素分泌或作用失调及β-细胞功能障碍,致使脂肪、碳水化合物以及蛋白质代谢紊乱,造成慢性血糖过高,最终导致各种血管及脏器并发症的出现。
3.糖尿病是一种进展型疾病,当生活干预后,血糖控制无法达标时,需要进行药物干预治疗。自1920年人类使用动物胰岛素治疗糖尿病以来,在100年的时间里,全球共发现、发明了九大类主要的糖尿病治疗药物:双胍类、磺脲类、噻唑烷二酮类(tzds)、格列奈类、α-糖苷酶抑制剂类、dpp-4抑制剂类、sglt-2抑制剂类、胰岛素类、glp-1类。其中,glp-1于1985年被发现,其是在人体进食后由胰高血糖素原基因表达,并主要在肠道黏膜l细胞分泌的一种产物,它能刺激胰岛β-细胞分泌胰岛素,对稳定血糖水平有重要作用。但glp-1在体内很容易被dpp4降解,半衰期不足2分钟,几乎不能成为有效的抗糖尿病药物。
4.艾塞那肽(exenatide,或称exendin-4)是在爬行动物毒蜥的唾液分泌物中发现的多肽,与glp-1(7-36)具有高度同源性(53%)。研究表明,exendin-4同样可以与glp-1受体结合,并在药理上表现出与glp-1类似的激动作用,例如增加胰岛素的合成及葡萄糖依赖性促胰岛素分泌;刺激β-细胞增生和再生,抑制β-细胞凋亡,从而增加β-细胞的数量;抑制胰高血糖素的分泌;抑制肝糖生成,但不会引起严重低血糖;抑制餐后胃肠道动力及分泌功能;降低食欲,减少食物的摄入;对神经细胞具有保护作用。exendin-4促进胰岛素分泌和抑制餐后胰高血糖素分泌的作用具有血糖依赖性,优于目前所用的磺酰脲类降糖药,不易发生低血糖反应,能够极大减少血糖监测次数,并能减轻体重。
5.聚合物修饰技术是上世纪70年代发展起来的一种强有力的修饰技术,其中尤以聚乙二醇化(pegylation)技术为代表。该技术是将聚乙二醇(peg)与蛋白质药物进行化学结合,对蛋白质的表面进行修饰。通过peg的修饰,一方面增加了蛋白质的分子量,降低了其在肾脏的排泄速率;另一方面,偶联的peg链在被修饰的蛋白质分子表面产生空间位阻效应,减低血液中蛋白水解酶对该蛋白的水解作用,从而有效地延长了其在循环系统内的滞留时间,导致药物血浆周期延长和系统性药物暴露增加并提高疗效。
6.相比于艾塞那肽或其变体,经特异性修饰而得到其聚乙二醇缀合物在作用持续时
间、生物利用度及治疗效果等方面均具有明显优势。临床研究表明,聚乙二醇化艾塞那肽或其变体对于ii型糖尿病患者具有优异的疗效和安全性。然而,目前尚无适合大规模生产的聚乙二醇化艾塞那肽或其变体的合成及纯化工艺。
技术实现要素:
7.发明要解决的问题
8.鉴于目前尚无适合大规模生产的聚乙二醇化艾塞那肽或其变体的合成及纯化工艺,本发明开发出一种用于制备特定艾塞那肽变体的聚乙二醇缀合物的方法,同时本发明还相应开发出一种用于制备作为原料使用的特定艾塞那肽变体的方法。
9.用于解决问题的方案
10.一方面,本发明提供了一种艾塞那肽变体cys
39-exendin-4的制备方法,其包括以下步骤:采用三步反相柱色谱法纯化cys
39-exendin-4粗品。
11.可选地,所述cys
39-exendin-4粗品通过固相合成方法得到。
12.可选地,所述三步反相柱色谱法中的每一步使用的色谱柱各自独立地选自丁烷基硅烷键合硅胶色谱柱、辛烷基硅烷键合硅胶色谱柱和十八烷基硅烷键合硅胶色谱柱中的一种或多种,优选十八烷基硅烷键合硅胶色谱柱。
13.可选地,所述三步反相柱色谱法中的第一步反相柱色谱法使用二元流动相体系,其中流动相a为酸性物质在水中的溶液,流动相b为酸性物质在有机溶剂中的溶液;流动相a和流动相b中的酸性物质各自独立地选自甲酸、乙酸、三氟乙酸、磷酸、磷酸氢二钾和磷酸二氢钾中的一种或多种,优选三氟乙酸;有机溶剂选自甲醇、乙醇和乙腈中的一种或多种,优选乙腈。进一步地,所述第一步反相柱色谱法使用的流动相a和流动相b中的酸性物质的体积百分比各自独立地为0.01%-1%,优选0.02-0.5%,更优选0.05-0.2%,甚至更优选0.1%。
14.可选地,所述第一步反相柱色谱法包括使用体积百分比为0%-95%,优选1%-90%,更优选5%-85%,甚至更优选8%-80%的流动相b进行的梯度洗脱过程。进一步地,所述梯度洗脱过程的时间为120min以内,优选110min以内,更优选100min以内,甚至更优选90min以内。
15.可选地,所述第一步反相柱色谱法还包括在梯度洗脱过程结束后进行的、流动相b的体积百分比与梯度终点相同的等度洗脱过程,优选还包括在梯度洗脱过程开始前和/或等度洗脱过程结束后进行的、流动相b的体积百分比与梯度起点相同的色谱柱平衡过程。
16.可选地,所述第一步反相柱色谱法的过程为:首先使用体积百分比为8%的流动相b进行色谱柱平衡,其次在90min内使用体积百分比为8%-80%的流动相b进行梯度洗脱,再次使用体积百分比为80%的流动相b进行等度洗脱,最后使用体积百分比降至8%的流动相b进行色谱柱再平衡。
17.可选地,所述三步反相柱色谱法中的第二步反相柱色谱法使用二元流动相体系,其中流动相a为酸性物质在水中的溶液,流动相b为有机溶剂;流动相a中的酸性物质选自磷酸、磷酸氢二钾、磷酸二氢钾、磷酸氢二钠和磷酸二氢钠中的一种或多种,优选磷酸二氢钾和磷酸;有机溶剂选自甲醇、乙醇和乙腈中的一种或多种,优选乙腈。进一步地,所述第二步反相柱色谱法使用的流动相a的ph值为1.0-6.0,优选2.0-5.0,更优选2.5-3.5,甚至更优选
3.0。
18.可选地,所述第二步反相柱色谱法包括使用体积百分比为0%-85%,优选1%-75%,更优选2%-70%,甚至更优选5%-65%的流动相b进行的梯度洗脱过程。进一步地,所述梯度洗脱过程的时间为100min以内,优选90min以内,更优选80min以内,甚至更优选70min以内。
19.可选地,所述第二步反相柱色谱法还包括在梯度洗脱过程结束后进行的、流动相b的体积百分比与梯度终点相同的等度洗脱过程,优选还包括在梯度洗脱过程开始前和/或等度洗脱过程结束后进行的、流动相b的体积百分比与梯度起点相同的色谱柱平衡过程。
20.可选地,所述第二步反相柱色谱法的过程为:首先使用体积百分比为5%的流动相b进行色谱柱平衡,其次在70min内使用体积百分比为5%-65%的流动相b进行梯度洗脱,再次使用体积百分比为65%的流动相b进行等度洗脱,最后使用体积百分比降至5%的流动相b进行色谱柱再平衡。
21.可选地,所述三步反相柱色谱法中的第三步反相柱色谱法使用二元流动相体系,其中流动相a为酸性物质在水中的溶液,流动相b为酸性物质在有机溶剂中的溶液;流动相a和流动相b中的酸性物质各自独立地选自甲酸、乙酸、三氟乙酸、磷酸、磷酸氢二钾和磷酸二氢钾中的一种或多种,优选三氟乙酸;有机溶剂选自甲醇、乙醇和乙腈中的一种或多种,优选乙腈。进一步地,所述第三步反相柱色谱法使用的流动相a和流动相b中的酸性物质的体积百分比各自独立地为0.005%-0.5%,优选0.01-0.2%,更优选0.02-0.1%,甚至更优选0.02%。
22.可选地,所述第三步反相柱色谱法包括使用体积百分比为0%-95%,优选1%-90%,更优选2%-85%,甚至更优选5%-80%的流动相b进行的梯度洗脱过程。进一步地,所述梯度洗脱过程的时间为100min以内,优选90min以内,更优选80min以内,甚至更优选75min。
23.可选地,所述第三步反相柱色谱法还包括在梯度洗脱过程结束后进行的、流动相b的体积百分比与梯度终点相同的等度洗脱过程,优选还包括在梯度洗脱过程开始前和/或等度洗脱过程结束后进行的、流动相b的体积百分比与梯度起点相同的色谱柱平衡过程。
24.可选地,所述第三步反相柱色谱法的过程为:首先使用体积百分比为5%的流动相b进行色谱柱平衡,其次在75min内使用体积百分比为5%-80%的流动相b进行梯度洗脱,再次使用体积百分比为80%的流动相b进行等度洗脱,最后使用体积百分比降至5%的流动相b进行色谱柱再平衡。
25.另一方面,本发明提供了一种聚乙二醇化艾塞那肽变体mpeg-ppmal-cys
39-exendin-4的制备方法,其包括以下步骤:
26.先通过本发明的艾塞那肽变体cys
39-exendin-4的制备方法得到cys
39-exendin-4,再将其与mpeg-ppmal进行缀合并纯化。
27.可选地,所述mpeg-ppmal的分子量为5-100kda,优选10-50kda,更优选15-40kda,甚至更优选20-29kda,甚至再优选23kda。
28.再一方面,本发明提供了一种聚乙二醇化艾塞那肽变体mpeg-ppmal-cys
39-exendin-4的制备方法,其包括以下步骤:
29.先将纯化后的cys
39-exendin-4与mpeg-ppmal进行缀合,再依次采用离子交换色谱
法与三步反相柱色谱法纯化mpeg-ppmal-cys
39-exendin-4粗品。
30.可选地,所述mpeg-ppmal的分子量为5-100kda,优选10-50kda,更优选15-40kda,甚至更优选20-29kda,甚至再优选23kda。
31.可选地,所述纯化后的cys
39-exendin-4通过本发明的艾塞那肽变体cys
39-exendin-4的制备方法得到。
32.可选地,所述离子交换色谱法为阳离子交换色谱法,使用的固定相为macrocap sp。
33.可选地,所述离子交换色谱法使用的流动相体系包含流动相a、流动相b、流动相c和流动相d;流动相a为乙酸和乙酸钠在水中的溶液;流动相a的ph值为2.0-6.0,优选3.0-5.0,更优选4.0;流动相a中的乙酸钠的浓度为5-50mmol/l,优选10-30mmol/l,更优选20mmol/l;流动相b为乙酸、乙酸钠和氯化钠在水中的溶液;流动相b的ph值为2.0-6.0,优选3.0-5.0,更优选4.0;流动相b中的乙酸钠的浓度为5-50mmol/l,优选10-30mmol/l,更优选20mmol/l;流动相b中的氯化钠的浓度为0.1-5mol/l,优选0.2-4mol/l,更优选0.5-2mol/l,甚至更优选1mol/l;流动相c为流动相a和流动相b的混合物,流动相a与流动相b的体积比为0.5:1-10:1,优选1:1-5:1,更优选2:1-4:1,甚至更优选4:1;流动相d为氢氧化钠在水中的溶液,流动相d中的氢氧化钠的浓度为0.02-2mol/l,优选0.05-1mol/l,更优选0.1-0.5mol/l,甚至更优选0.2mol/l。
34.可选地,所述离子交换色谱法的过程为:首先依次使用流动相d、流动相b和流动相a冲洗akta系统,其次使用流动相a稀释预先配制的聚乙二醇化艾塞那肽变体mpeg-ppmal-cys
39-exendin-4粗品溶液后上样,最后依次使用流动相a和流动相c冲洗akta系统,并收集组分。
35.可选地,所述三步反相柱色谱法中的每一步使用的色谱柱各自独立地选自丁烷基硅烷键合硅胶色谱柱(c4柱)、辛烷基硅烷键合硅胶色谱柱(c8柱)和十八烷基硅烷键合硅胶色谱柱(c18柱)中的一种或多种,优选十八烷基硅烷键合硅胶色谱柱(c18柱)。
36.可选地,所述三步反相柱色谱法中的第一步和第三步反相柱色谱法使用相同的二元流动相体系和洗脱过程,其中流动相a为酸性物质在水中的溶液,流动相b为酸性物质在有机溶剂中的溶液;流动相a和流动相b中的酸性物质各自独立地选自甲酸、乙酸、三氟乙酸、磷酸、磷酸氢二钾和磷酸二氢钾中的一种或多种,优选三氟乙酸;有机溶剂选自甲醇、乙醇和乙腈中的一种或多种,优选乙腈。进一步地,所述第一步和第三步反相柱色谱法使用的流动相a和流动相b中的酸性物质的体积百分比各自独立地为0.01%-1%,优选0.02-0.5%,更优选0.05-0.2%,甚至更优选0.1%。
37.可选地,所述第一步和第三步反相柱色谱法包括使用体积百分比为0%-95%,优选2%-90%,更优选5%-85%,甚至更优选9%-80%的流动相b进行的梯度洗脱过程。进一步地,所述梯度洗脱过程的时间为100min以内,优选90min以内,更优选80min以内,甚至更优选70min以内。
38.可选地,所述三步反相柱色谱法中的第二步反相柱色谱法使用二元流动相体系,其中流动相a为酸性物质在水中的溶液,流动相b为有机溶剂;流动相a中的酸性物质选自磷酸、磷酸氢二钾、磷酸二氢钾、磷酸氢二钠和磷酸二氢钠中的一种或多种,优选磷酸二氢钾和磷酸;有机溶剂选自甲醇、乙醇和乙腈中的一种或多种,优选乙腈。进一步地,所述第二步
反相柱色谱法使用的流动相a的ph值为1.0-6.0,优选2.0-5.0,更优选2.5-3.5,甚至更优选3.0。
39.可选地,所述第二步反相柱色谱法包括使用体积百分比为0%-85%,优选2%-75%,更优选5%-70%,甚至更优选8%-65%的流动相b进行的梯度洗脱过程。进一步地,所述梯度洗脱过程的时间为100min以内,优选90min以内,更优选80min以内,甚至更优选70min以内。
40.可选地,所述制备方法还包括在三步反相柱色谱法结束后进行的超滤过程。
41.可选地,所述超滤过程为:首先将通过三步反相柱色谱法纯化后得到的样品进行超滤,其次使用水进行稀释,再次使用ph调节剂将ph值调节至4.0-5.0,最后再次进行超滤;ph调节剂为碱性物质,优选碱金属的氢氧化物、碳酸盐和碳酸氢盐中的一种或多种,更优选氢氧化钠、碳酸钠、碳酸氢钠、氢氧化钾、碳酸钾和碳酸氢钾中的一种或多种,甚至更优选碳酸氢钠。
42.发明的效果
43.为了弥补聚乙二醇化艾塞那肽或其变体的大规模生产工艺的空白,本发明相应开发出cys
39-exendin-4多肽和mpeg-ppmal-cys
39-exendin-4缀合物的制备方法。在纯化cys
39-exendin-4多肽的过程中,本发明的多肽制备方法创造性地使用了三步反相柱色谱法,并且进一步优化了第二步反相柱色谱法使用的酸性物质,使得目标产品的纯度(可达98%以上)和产量均有明显提高。在纯化mpeg-ppmal-cys
39-exendin-4缀合物的过程中,本发明的缀合物制备方法创造性地依次使用了离子交换色谱法和三步反相柱色谱法,使得目标产品的纯度可达99.5%以上,并且产量较高。组合使用本发明中的多肽和缀合物两种制备方法,基本上能够满足工业化生产的需要。
具体实施方式
44.术语定义
45.除非另有说明,本发明上下文中使用的术语“色谱法”(或称“层析法”)意指一种分离分析方法,其利用各组分的物理化学性质差异,在流动相(洗脱剂)和固定相(填料)之间因吸附、分配或亲和力的不同而产生差速迁移,从而达到分离的目的,并随后借助分析手段达到分析的目的。色谱法是分离分析混合物最有力的手段,具有高灵敏度、高选择性、高效能、分析速度快及应用范围广等优点。
46.除非另有说明,本发明上下文中使用的术语“固定相”意指在色谱分离过程中固定不动、对样品产生保留作用的一相。固定相的选择对样品的分离发挥重要作用,有时甚至是决定性的作用。不同类型的色谱法采用不同的固定相,气-固色谱法的固定相为具有吸附活性的固体吸附剂,例如聚硅氧烷、聚乙二醇、氧化铝等;气-液色谱法的固定相为载体表面涂渍的固定液;液相色谱法的固定相为各种键合型的硅胶小球,例如c4柱中的丁烷基硅烷键合硅胶bs、c8柱中的辛烷基硅烷键合硅胶os、c18柱中的十八烷基硅烷键合硅胶ods等;离子交换色谱法的固定相为各种离子交换剂;空间排阻色谱法的固定相为各种不同类型的凝胶等。
47.除非另有说明,本发明上下文中使用的术语“流动相”意指在色谱分离过程中与固定相处于平衡状态、带动样品向前移动的另一相。流动相的选择对样品的分离同样发挥重
要作用。不同类型的色谱法通常不同的流动相,气相色谱法的流动相(或称“载气”)通常为高纯氦气;液相色谱法的流动相则相对复杂且灵活,既可以使用一元流动相体系(例如水或其缓冲盐溶液、甲醇、乙腈等),也可以使用二元流动相体系(例如乙腈-三氟乙酸水溶液、三氟乙酸乙腈溶液-三氟乙酸水溶液、乙腈-磷酸盐缓冲液、乙腈-乙酸盐缓冲液等)。
48.当使用二元流动相体系时,通常会采用梯度洗脱(或称梯度淋洗或程序洗脱)的洗脱方式。除非另有说明,本发明上下文中使用的术语“梯度洗脱”意指在一个分析周期内,按照一定程序不断改变两种流动相的浓度或体积百分比的配比。梯度洗脱可以使一个复杂样品中性质差异较大的组分都能在各自适宜的分离条件下分离。
49.色谱法可从不同的角度进行分类:
50.1.按照流动相与固定相的分子聚集状态:在色谱法中,流动相可以是气体、液体和超临界流体,分别对应气相色谱法(gc;按照固定相的不同,还可细分为气-固色谱法gsc和气-液色谱法glc)、液相色谱法(lc;按照固定相的不同,还可细分为液-固色谱法lsc和液-液色谱法llc)和超临界流体色谱法(sfc)。
51.2.按照操作形式:可分为柱色谱法(cc;按照色谱柱规格的不同,还可细分填充柱色谱法pc、微填充柱色谱法mpc和毛细管柱色谱法cc;在液相柱色谱法中,按照分离能力的不同,还可细分为经典液相柱色谱法lcc(即狭义上的柱色谱法)、高效液相色谱法hplc和超高效液相色谱法uplc等)、平板色谱法(pc;按照固定相载体的不同,还可细分为利用滤纸的纸色谱法pc、利用玻璃板或铝箔板薄层色谱法tlc和利用高分子膜的薄膜色谱法tfc)、毛细管电泳法(ce)等。
52.除非另有说明,本发明上下文中使用的术语“高效液相色谱法”意指相对于经典液相柱色谱法,流动相改为高压输送、采用高效固定相、具有在线检测器及仪器化的液相色谱法,具有分离效能高、分析速度快及应用范围广等特点,因此又被称为高速液相色谱法(hslc)、高压液相色谱法(hplc)、高分辨液相色谱法(hrlc)等
53.3.按照分离机制:可分为分配色谱法(pc)、吸附色谱法(ac)、离子交换色谱法(iec)、空间排阻色谱法(sec)和亲和色谱法(ac)等。
54.除非另有说明,本发明上下文中使用的术语“离子交换色谱法”意指利用被分离组分离子交换能力的差别而实现分离的方法,其固定相为离子交换树脂,按照可交换离子的电荷符号的不同,还可细分为阳离子交换树脂和阴离子交换树脂,分别对应阳离子交换色谱法(cec)和阴离子交换色谱法(aec)。
55.4.按照固定相和流动相的极性强弱:可分为正相色谱法(np-c)和反相色谱法(rp-c),其中固定相的极性大于流动相,则为正相色谱法;流动相的极性大于固定相,则为反相色谱法;相应地,高效液相色谱法(hplc)也可分为正相高效液相色谱法(np-hplc)和反相高效液相色谱法(rp-hplc)。
56.除非另有说明,本发明上下文中使用的术语“超滤”意指以压力为推动力,以实现大分子与小分子的分离为目的的膜分离技术。在一定压力下,小分子溶质和溶剂可以穿过一定孔径的分离用薄膜,而大分子溶质无法穿过,从而使大分子物质得到一定程度的纯化。超滤技术可以用于分离蛋白质、酶、核酸、多糖、多肽、抗生素、病毒等,整个分离过程中没有相转移,无需添加任何强烈化学物质,可以在低温下操作,过滤速率较快,便于进行无菌处理,避免了生物活性物质的活力损失和变性。
57.除非另有说明,本发明上下文中使用的术语“固相合成”意指一种将反应物连接在反应溶剂不溶性的固相载体上进行的合成方法,多用于多肽等物质的合成,即“固相多肽合成”(spps)。首先,将一个氨基被保护基封闭的氨基酸共价连接在固相载体上,再脱掉氨基的保护基,从而完成第一个氨基酸与固相载体的连接;然后,氨基被封闭的第二个氨基酸的羧基通过缩合剂被活化,再与已接在固相载体上的第一个氨基酸的氨基形成肽键,在固相载体上得到带有保护基的二肽;重复上述形成肽键的反应,使肽链从c端向n端生长,直至达到所需的肽链长度;最后,脱去n段氨基酸的氨基保护基,使用裂解液将多肽c端从固相载体上剥离,就得到了目标多肽。固相多肽合成的优点主要表现在最初的反应物和产物都连接在固相载体上,因此可以在一个反应容器中进行所有的反应,便于自动化操作,加入过量的反应物可以获得高产率的产物,同时产物比较容易分离。目前,固相多肽合成可以在程序控制的自动化多肽合成仪上进行。
58.除非另有说明,本发明上下文中使用的术语“艾塞那肽变体”意指cys
39-exendin-4(或cys
39-exenatide),其为艾塞那肽氨基酸序列中第39位的丝氨酸(ser)突变为半胱氨酸(cys)后得到的变体。具体地,cys
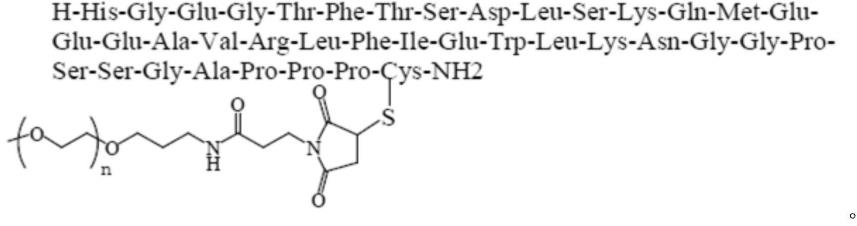
39-exendin-4的结构为:h-his-gly-glu-gly-thr-phe-thr-ser-asp-leu-ser-lys-gln-met-glu-glu-glu-ala-val-arg-leu-phe-ile-glu-trp-leu-lys-asn-gly-gly-pro-ser-ser-gly-ala-pro-pro-pro-cys-nh2,如seq id no:1所示。
59.除非另有说明,本发明上下文中使用的术语“聚乙二醇化艾塞那肽变体”意指mpeg-ppmal-cys
39-exendin-4(或mpeg-ppmal-cys
39-exenatide),其为cys
39-exenatide与α-甲氧基-聚乙二醇-ω-[3-(3-马来酰亚胺基-1-氧代丙基)氨基]丙基醚(mpeg-ppmal)的缀合物,可简称为“艾塞那肽变体-聚乙二醇缀合物”。具体地,mpeg-ppmal-cys
39-exendin-4的结构为:
[0060][0061]
上述聚乙二醇化艾塞那肽变体中的聚乙二醇修饰剂部分的分子量可以为5-100kda,优选10-50kda,更优选15-40kda,例如15kda、16kda、17kda、18kda、19kda、20kda、21kda、22kda、23kda、24kda、25kda、26kda、27kda、28kda、29kda、30kda、31kda、32kda、33kda、34kda、35kda、36kda、37kda、38kda、39kda、40kda,甚至更优选20-29kda,甚至再优选23kda。
[0062]
由于聚合物是由一定分布范围内的不同聚合度的分子构成,一般用平均分子量表示聚合物的分子量。具体来说,可以是数均分子量,或重均分子量。尽管在聚合物的聚合度差异较大时,数均分子量和重均分子量可能有一些偏差,但是对于分布范围较窄的聚合物来说,二者趋于相等。对于本发明的聚乙二醇缀合物,在提及其分子量时,既可以是重均分子量,也可以是数均分子量,优选数均分子量。
[0063]
除非另有说明,本发明上下文中使用的术语“缀合物”意指将蛋白质、多肽、糖、脂
质、核酸等大分子化合物与聚乙二醇修饰剂等小分子和/或其他大分子化合物以共价键相互连结而形成的化合物;相应地,缀合物中包含的不同片段之间形成共价键的过程即为“缀合”。除非另有说明,本发明上下文中使用的术语“聚乙二醇修饰剂”(或称“peg修饰剂”意指带有可与待修饰分子反应的官能团的聚乙二醇衍生物(例如mpeg-nh2、mpeg-ots、mpeg-ppmal等),多用于多肽或蛋白质类药物的结构修饰,以增加体内半衰期,降低免疫原性,同时还可以增加药物的水溶性等。
[0064]
艾塞那肽变体(cys
39-exendin-4)的制备方法
[0065]
针对上述艾塞那肽变体,本发明提供了一种cys
39-exendin-4的制备方法,其可以包括以下步骤:采用三步反相柱色谱法纯化cys
39-exendin-4粗品。
[0066]
在本发明的一项实施方案中,上述多肽制备方法中的cys
39-exendin-4粗品可以通过固相合成方法得到。
[0067]
在本发明的一项实施方案中,上述多肽制备方法中的cys
39-exendin-4粗品可以通过固相多肽合成方法得到。
[0068]
上述固相合成方法或固相多肽合成方法的具体过程可以由所属领域技术人员根据实际需要而确定,例如以fmoc保护的氨基树脂为载体,采用fmoc保护的氨基酸依序连接的合成方法。
[0069]
上述多肽制备方法中的纯化步骤可以包括三步反相柱色谱法,每一步使用的色谱柱可以各自独立地进行选择,彼此之间既可以完全相同,也可以不尽相同,还可以完全不同,例如各自独立地选自c4柱、c8柱和c18柱中的一种或多种。
[0070]
在本发明的一项实施方案中,上述多肽制备方法中的纯化步骤可以包括三步反相柱色谱法(如rp-hplc,特别是制备型rp-hplc),三步使用的色谱柱可以均为c18柱。
[0071]
上述三步反相柱色谱法中的第一步反相柱色谱法可以使用二元流动相体系,其中流动相a可以为酸性物质在水中的溶液,流动相b可以为酸性物质在有机溶剂中的溶液;流动相a和流动相b中的酸性物质可以各自独立地进行选择,彼此之间既可以相同,也可以不同,例如各自独立地选自甲酸、乙酸、三氟乙酸、磷酸、磷酸氢二钾和磷酸二氢钾中的一种或多种,优选三氟乙酸;有机溶剂可以为本领域的常用有机溶剂,例如选自甲醇、乙醇和乙腈中的一种或多种,优选乙腈。
[0072]
上述三步反相柱色谱法中的第一步反相柱色谱法使用的流动相a和流动相b中的酸性物质的体积百分比既可以相同,也可以不同,例如各自独立地为0.01%-1%,优选0.02-0.5%,更优选0.05-0.2%,甚至更优选0.1%。
[0073]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第一步反相柱色谱法可以使用二元流动相体系,其中流动相a可以为三氟乙酸的水溶液,流动相b可以为三氟乙酸的乙腈溶液;流动相a和流动相b中的三氟乙酸的体积百分比可以均为0.1%。
[0074]
上述三步反相柱色谱法中的第一步反相柱色谱法可以包括使用体积百分比为0%-95%,优选1%-90%,更优选5%-85%,甚至更优选8%-80%的流动相b进行的梯度洗脱过程;梯度洗脱过程的时间可以为120min以内,优选110min以内,更优选100min以内,甚至更优选90min以内。
[0075]
上述三步反相柱色谱法中的第一步反相柱色谱法还可以包括在梯度洗脱过程结束后进行的、流动相b的体积百分比与梯度终点相同的等度洗脱过程,优选还可以包括在梯
度洗脱过程开始前和/或等度洗脱过程结束后进行的、流动相b的体积百分比与梯度起点相同的色谱柱平衡过程。
[0076]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第一步反相柱色谱法的过程可以为:首先使用体积百分比为8%的流动相b进行色谱柱平衡,其次在90min内使用体积百分比为8%-80%的流动相b进行梯度洗脱,再次使用体积百分比为80%的流动相b进行等度洗脱,最后使用体积百分比降至8%的流动相b进行色谱柱再平衡。
[0077]
上述三步反相柱色谱法中的第二步反相柱色谱法可以使用二元流动相体系,其中流动相a可以为酸性物质在水中的溶液,流动相b可以为有机溶剂;流动相a中的酸性物质可以选自磷酸、磷酸氢二钾、磷酸二氢钾、磷酸氢二钠和磷酸二氢钠的一种或多种,优选磷酸二氢钾和磷酸;有机溶剂可以为本领域的常用有机溶剂,例如选自甲醇、乙醇和乙腈中的一种或多种,优选乙腈。
[0078]
上述三步反相柱色谱法中的第二步反相柱色谱法使用的流动相a的ph值可以为1.0-6.0,优选2.0-5.0,更优选2.5-3.5,甚至更优选3.0。
[0079]
当流动相a中包含磷酸盐时,所述磷酸盐的浓度可以为5-100mmol/l,优选10-80mmol/l,更优选15-70mmol/l,甚至更优选20-50mmol/l。
[0080]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第二步反相柱色谱法可以使用二元流动相体系,其中流动相a可以为磷酸二氢钾和磷酸的水溶液,磷酸二氢钾的浓度可以为35mmol/l,流动相a的ph值可以为3.0,流动相b可以为乙腈。
[0081]
上述三步反相柱色谱法中的第二步反相柱色谱法可以包括使用体积百分比为0%-85%,优选1%-75%,更优选2%-70%,甚至更优选5%-65%的流动相b进行的梯度洗脱过程;梯度洗脱过程的时间可以为100min以内,优选90min以内,更优选80min以内,甚至更优选70min以内。
[0082]
上述三步反相柱色谱法中的第二步反相柱色谱法还可以包括在梯度洗脱过程结束后进行的、流动相b的体积百分比与梯度终点相同的等度洗脱过程,优选还可以包括在梯度洗脱过程开始前和/或等度洗脱过程结束后进行的、流动相b的体积百分比与梯度起点相同的色谱柱平衡过程。
[0083]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第二步反相柱色谱法的过程可以为:首先使用体积百分比为5%的流动相b进行色谱柱平衡,其次在70min内使用体积百分比为5%-65%的流动相b进行梯度洗脱,再次使用体积百分比为65%的流动相b进行等度洗脱,最后使用体积百分比降至5%的流动相b进行色谱柱再平衡。
[0084]
上述三步反相柱色谱法中的第三步反相柱色谱法可以使用二元流动相体系,其中流动相a可以为酸性物质在水中的溶液,流动相b可以为酸性物质在有机溶剂中的溶液;水和有机溶剂中的酸性物质可以各自独立地进行选择,彼此之间既可以相同,也可以不同,例如各自独立地选自甲酸、乙酸、三氟乙酸、磷酸、磷酸氢二钾和磷酸二氢钾中的一种或多种,优选三氟乙酸;有机溶剂可以为本领域的常用有机溶剂,例如选自甲醇、乙醇和乙腈中的一种或多种,优选乙腈。
[0085]
上述三步反相柱色谱法中的第三步反相柱色谱法使用的流动相a和流动相b中的酸性物质的体积百分比既可以相同,也可以不同,例如各自独立地为0.005%-0.5%,优选0.01-0.2%,更优选0.02-0.1%,甚至更优选0.02%。
[0086]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第三步反相柱色谱法可以使用二元流动相体系,其中流动相a可以为三氟乙酸的水溶液,流动相b可以为三氟乙酸的乙腈溶液;流动相a和流动相b中的三氟乙酸的体积百分比可以均为0.02%。
[0087]
上述三步反相柱色谱法中的第三步反相柱色谱法可以包括使用体积百分比为0%-95%,优选1%-90%,更优选2%-85%,甚至更优选5%-80%的流动相b进行的梯度洗脱过程;梯度洗脱过程的时间为100min以内,优选90min以内,更优选80min以内,甚至更优选75min。
[0088]
上述三步反相柱色谱法中的第三步反相柱色谱法还可以包括在梯度洗脱过程结束后进行的、流动相b的体积百分比与梯度终点相同的等度洗脱过程,优选还包括在梯度洗脱过程开始前和/或等度洗脱过程结束后进行的、流动相b的体积百分比与梯度起点相同的色谱柱平衡过程。
[0089]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第三步反相柱色谱法的过程可以为:首先使用体积百分比为5%的流动相b进行色谱柱平衡,其次在75min内使用体积百分比为5%-80%的流动相b进行梯度洗脱,再次使用体积百分比为80%的流动相b进行等度洗脱,最后使用体积百分比降至5%的流动相b进行色谱柱再平衡。
[0090]
聚乙二醇化艾塞那肽变体(mpeg-ppmal-cys
39-exendin-4)的制备方法
[0091]
针对上述聚乙二醇化艾塞那肽变体,本发明提供了一种mpeg-ppmal-cys
39-exendin-4的制备方法,其可以包括以下步骤:先通过本发明的多肽制备方法得到cys
39-exendin-4,再将其与mpeg-ppmal进行缀合并纯化。
[0092]
在本发明的一项实施方案中,上述缀合物制备方法中的mpeg-ppmal的分子量(例如数均分子量)可以为5-100kda。
[0093]
在本发明的一项实施方案中,上述缀合物制备方法中的mpeg-ppmal的分子量(例如数均分子量)可以为10-50kda。
[0094]
在本发明的一项实施方案中,上述缀合物制备方法中的mpeg-ppmal的分子量(例如数均分子量)可以为15-40kda。
[0095]
在本发明的一项实施方案中,上述缀合物制备方法中的mpeg-ppmal的分子量(例如数均分子量)可以为20-29kda。
[0096]
在本发明的一项实施方案中,上述缀合物制备方法中的mpeg-ppmal的分子量(例如数均分子量)可以为23kda。
[0097]
上述缀合物制备方法中缀合步骤的具体过程可以由所属领域技术人员根据实际需要而确定,例如通过加成反应(如迈克尔加成反应)来完成。
[0098]
上述缀合物制备方法中纯化步骤的具体过程可以由所属领域技术人员根据实际需要而确定,例如通过离子交换色谱法(如cec)和反相柱色谱法(如rp-hplc,特别是制备型rp-hplc)中的一种或多种来完成。
[0099]
针对上述聚乙二醇化艾塞那肽变体,本发明还提供了另一种mpeg-ppmal-cys
39-exendin-4的制备方法,其可以包括以下步骤:先将纯化后的cys
39-exendin-4与mpeg-ppmal进行缀合,再依次采用离子交换色谱法与三步反相柱色谱法纯化mpeg-ppmal-cys
39-exendin-4粗品。
[0100]
在本发明的一项实施方案中,上述缀合物制备方法中的mpeg-ppmal的分子量(例
如数均分子量)可以为5-100kda。
[0101]
在本发明的一项实施方案中,上述缀合物制备方法中的mpeg-ppmal的分子量(例如数均分子量)可以为10-50kda。
[0102]
在本发明的一项实施方案中,上述缀合物制备方法中的mpeg-ppmal的分子量(例如数均分子量)可以为15-40kda。
[0103]
在本发明的一项实施方案中,上述缀合物制备方法中的mpeg-ppmal的分子量(例如数均分子量)可以为20-29kda。
[0104]
在本发明的一项实施方案中,上述缀合物制备方法中的mpeg-ppmal的分子量(例如数均分子量)可以为23kda。
[0105]
上述缀合物制备方法中缀合步骤的具体过程可以由所属领域技术人员根据实际需要而确定,例如cys
39-exenatide通过其第39位cys残基中的巯基(-sh)与mpeg-ppmal发生迈克尔加成反应(michael addition reaction)而得到。
[0106]
在本发明的一项实施方案中,上述缀合物制备方法中的纯化后的cys
39-exendin-4可以通过本发明的多肽制备方法得到。
[0107]
在本发明的一项实施方案中,上述缀合物制备方法中的离子交换色谱法可以为cec,例如使用macrocap sp作为固定相。
[0108]
上述缀合物制备方法中的离子交换色谱法使用的流动相体系可以包含流动相a、流动相b、流动相c和流动相d。
[0109]
上述离子交换色谱法使用的流动相体系中的流动相a可以为乙酸和乙酸钠在水中的溶液;流动相a的ph值可以为2.0-6.0,优选3.0-5.0,更优选4.0;流动相a中的乙酸钠的浓度可以为5-50mmol/l,优选10-30mmol/l,更优选20mmol/l。
[0110]
在本发明的一项实施方案中,上述离子交换色谱法使用的流动相体系中的流动相a可以为乙酸和乙酸钠的水溶液;流动相a的ph值可以为4.0;流动相a中的乙酸钠的浓度可以为20mmol/l。
[0111]
上述离子交换色谱法使用的流动相体系中的流动相b可以为乙酸、乙酸钠和氯化钠在水中的溶液;流动相b的ph值可以为2.0-6.0,优选3.0-5.0,更优选4.0;流动相b中的乙酸钠的浓度可以为5-50mmol/l,优选10-30mmol/l,更优选20mmol/l;流动相b中的氯化钠的浓度可以为0.1-5mol/l,优选0.2-4mol/l,更优选0.5-2mol/l,甚至更优选1mol/l。
[0112]
在本发明的一项实施方案中,上述离子交换色谱法使用的流动相体系中的流动相b可以为乙酸、乙酸钠和氯化钠的水溶液;流动相b的ph值可以为4.0;流动相b中的乙酸钠的浓度可以为20mmol/l,氯化钠的浓度可以为1mol/l。
[0113]
上述离子交换色谱法使用的流动相体系中的流动相c可以为流动相a和流动相b的混合物,流动相a与流动相b的体积比可以为0.5:1-10:1,优选1:1-5:1,更优选2:1-4:1,甚至更优选4:1。
[0114]
在本发明的一项实施方案中,上述离子交换色谱法使用的流动相体系中的流动相c可以为流动相a和流动相b的混合物,流动相a与流动相b的体积比可以为4:1。
[0115]
上述离子交换色谱法使用的流动相体系中的流动相d可以为氢氧化钠在水中的溶液,流动相d中的氢氧化钠的浓度可以为0.02-2mol/l,优选0.05-1mol/l,更优选0.1-0.5mol/l,甚至更优选0.2mol/l。
[0116]
在本发明的一项实施方案中,上述离子交换色谱法使用的流动相体系中的流动相d可以为氢氧化钠的水溶液,流动相d中的氢氧化钠的浓度可以为0.2mol/l。
[0117]
在本发明的一项实施方案中,上述缀合物制备方法中的离子交换色谱法的过程可以为:首先依次使用流动相d、流动相b和流动相a冲洗akta系统,其次使用流动相a稀释预先配制的mpeg-ppmal-cys
39-exendin-4粗品溶液后上样,最后依次使用流动相a和流动相c冲洗akta系统,并收集组分。
[0118]
上述缀合物制备方法中的纯化步骤可以包括三步反相柱色谱法,每一步使用的色谱柱可以各自独立地进行选择,彼此之间既可以完全相同,也可以不尽相同,还可以完全不同,例如各自独立地选自c4柱、c8柱和c18柱中的一种或多种。
[0119]
在本发明的一项实施方案中,上述缀合物制备方法中的纯化步骤可以包括三步反相柱色谱法(如rp-hplc,特别是制备型rp-hplc),三步使用的色谱柱可以均为c18柱。
[0120]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第一步和第三步反相柱色谱法可以使用相同的二元流动相体系和洗脱过程,其中流动相a可以为酸性物质在水中的溶液,流动相b可以为酸性物质在有机溶剂中的溶液;流动相a和流动相b中的酸性物质可以各自独立地选自甲酸、乙酸、三氟乙酸、磷酸、磷酸氢二钾和磷酸二氢钾中的一种或多种,优选三氟乙酸;有机溶剂可以为本领域的常用有机溶剂,例如选自甲醇、乙醇和乙腈中的一种或多种,优选乙腈。
[0121]
上述三步反相柱色谱法中的第一步和第三步反相柱色谱法使用的流动相a和流动相b中的酸性物质的体积百分比既可以相同,也可以不同,例如各自独立地为0.01%-1%,优选0.02-0.5%,更优选0.05-0.2%,甚至更优选0.1%。
[0122]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第一步和第三步反相柱色谱法可以使用二元流动相体系,其中流动相a可以为三氟乙酸的水溶液,流动相b可以为三氟乙酸的乙腈溶液;流动相a和流动相b中的三氟乙酸的体积百分比可以均为0.1%。
[0123]
上述三步反相柱色谱法中的第一步和第三步反相柱色谱法可以包括使用体积百分比为0%-95%,优选2%-90%,更优选5%-85%,甚至更优选9%-80%的流动相b进行的梯度洗脱过程;梯度洗脱过程的时间可以为100min以内,优选90min以内,更优选80min以内,甚至更优选70min以内。
[0124]
上述三步反相柱色谱法中的第一步和第三步反相柱色谱法还可以包括在梯度洗脱过程结束后进行的、流动相b的体积百分比与梯度终点相同的等度洗脱过程,优选还可以包括在梯度洗脱过程开始前和/或等度洗脱过程结束后进行的、流动相b的体积百分比与梯度起点相同的色谱柱平衡过程。
[0125]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第一步和第三步反相柱色谱法的过程可以为:首先使用体积百分比为9%的流动相b进行色谱柱平衡,其次在70min内使用体积百分比为9%-80%的流动相b进行梯度洗脱,再次使用体积百分比为80%的流动相b进行等度洗脱,最后使用体积百分比降至9%的流动相b进行色谱柱再平衡。
[0126]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第二步反相柱色谱法可以使用二元流动相体系,其中流动相a可以为酸性物质在水中的溶液,流动相b可以为有机溶剂;流动相a中的酸性物质可以选自磷酸、磷酸氢二钾、磷酸二氢钾、磷酸氢二钠和磷酸二氢钠中的一种或多种,优选磷酸二氢钾和磷酸;有机溶剂可以为本领域的常用有机溶剂,例
如选自甲醇、乙醇和乙腈中的一种或多种,优选乙腈。
[0127]
上述三步反相柱色谱法中的第二步反相柱色谱法使用的流动相a的ph值可以为1.0-6.0,优选2.0-5.0,更优选2.5-3.5,甚至更优选3.0。
[0128]
当流动相a中包含磷酸盐(例如磷酸二氢钾)时,磷酸盐的浓度可以为5-100mmol/l,优选10-80mmol/l,更优选15-70mmol/l,甚至更优选20-50mmol/l。
[0129]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第二步反相柱色谱法可以使用二元流动相体系,其中流动相a可以为磷酸二氢钾和磷酸的水溶液,磷酸二氢钾的浓度可以为35mmol/l,流动相a的ph值可以为3.0,流动相b可以为乙腈。
[0130]
上述三步反相柱色谱法中的第二步反相柱色谱法可以包括使用体积百分比为0%-85%,优选2%-75%,更优选5%-70%,甚至更优选8%-65%的流动相b进行的梯度洗脱过程;梯度洗脱过程的时间可以为100min以内,优选90min以内,更优选80min以内,甚至更优选70min以内。
[0131]
上述三步反相柱色谱法中的第二步反相柱色谱法还可以包括在梯度洗脱过程结束后进行的、流动相b的体积百分比与梯度终点相同的等度洗脱过程,优选还可以包括在梯度洗脱过程开始前和/或等度洗脱过程结束后进行的、流动相b的体积百分比与梯度起点相同的色谱柱平衡过程。
[0132]
在本发明的一项实施方案中,上述三步反相柱色谱法中的第二步反相柱色谱法的过程可以为:首先使用体积百分比为8%的流动相b进行色谱柱平衡,其次在70min内使用体积百分比为8%-65%的流动相b进行梯度洗脱,再次使用体积百分比为65%的流动相b进行等度洗脱,最后使用体积百分比降至8%的流动相b进行色谱柱再平衡。
[0133]
上述缀合物制备方法中的纯化步骤还可以包括在三步反相柱色谱法结束后进行的超滤过程。
[0134]
上述缀合物制备方法中的超滤过程可以为:首先将通过三步反相柱色谱法纯化后得到的样品进行超滤,其次使用水进行稀释,再次使用ph调节剂将ph值调节至4.0-5.0,最后再次进行超滤;ph调节剂为碱性物质,优选碱金属的氢氧化物、碳酸盐和碳酸氢盐中的一种或多种,更优选氢氧化钠、碳酸钠、碳酸氢钠、氢氧化钾、碳酸钾和碳酸氢钾中的一种或多种,甚至更优选碳酸氢钠。
[0135]
在本发明的一项实施方案中,上述缀合物制备方法中的超滤过程可以为:首先将通过三步反相柱色谱法纯化后得到的样品进行超滤(如超滤4遍),其次使用水(如纯化水)进行稀释(如稀释1倍),再次使用碳酸氢钠(如0.1m水溶液)将ph值调节至4.0-5.0,最后再次进行超滤(如超滤3遍)。
[0136]
以下将结合具体实施例来进一步阐述本发明的技术方案。除非另有说明,下列实施例中使用的材料、试剂、仪器等均可通过常规商业手段获得。
[0137]
除非另有说明,本发明中以缩写表示的术语具有以下含义。
[0138]
术语缩写二氯甲烷dcmn,n-二异丙基乙胺dipean,n-二甲基甲酰胺dmf9-芴甲氧羰基fmoc
o-(苯并三唑-1-基)-n,n,n',n'-四甲基脲六氟磷酸盐hbtu氮气n2三氟乙酸tfa乙酸hac乙酸钠naac氯化钠nacl小时h分钟min柱体积cv
[0139]
实施例一:艾塞那肽变体cys
39-exendin-4的合成
[0140]
本发明中的艾塞那肽变体可以采用常规的固相合成方法得到。例如,使用以下的方法进行合成:
[0141]
(1)将fmoc-rink linker mbha树脂加入到合成仪反应罐中,加入dcm,开启搅拌,溶胀30min,之后向反应罐内充入n2,直至整个合成反应结束;
[0142]
(2)待树脂溶胀完毕后,排干溶液,根据树脂体积配制作为去保护剂的哌啶/dmf,将去保护剂加入反应罐内,与树脂搅拌反应,得到脱保护的树脂;
[0143]
(3)将fmoc保护的氨基酸以及作为缩合剂的hbtu/dipea溶解于dmf中,加入到反应罐内反应1-5h,用茚三酮方法监测缩合反应是否完全;
[0144]
(4)配制作为去保护剂的哌啶/dmf,将去保护剂加入反应罐内,反应10-30min,用茚三酮方法监测保护基是否完全脱除;
[0145]
(5)按照目标多肽的氨基酸序列,依次使用相应的fmoc保护的氨基酸,重复步骤(3)和(4),直至序列中最后的氨基酸偶联完毕;
[0146]
(6)将预先配制的tfa裂解液加入反应罐内,与偶联完毕的多肽树脂混合,反应3-5小时,过滤,向滤液中加入乙醚,静置后过滤,收集沉淀,得到艾塞那肽变体cys
39-exendin-4粗品。
[0147]
实施例二:艾塞那肽变体cys
39-exendin-4的纯化
[0148]
按照实施例一中记载的方法,制备三个批次的艾塞那肽变体cys
39-exendin-4粗品,每一批粗品均分成三份,分别使用40%v/v乙酸溶解,得到粗品溶液。
[0149]
分别采用下列纯化工艺(1)-(3)对每一批粗品中的三份样品进行纯化,然后收集样品,冻干,并进行相关检测:
[0150]
纯化工艺(1):方法a 方法c 方法d;
[0151]
纯化工艺(2):方法a 方法d;
[0152]
纯化工艺(3):方法a 方法b 方法d。
[0153]
上述纯化工艺中使用的方法a-d的具体情况如下:
[0154]
方法a:rp-hplc
[0155]
色谱柱填料:c18 smb200-10;
[0156]
检测波长:230nm;
[0157]
流动相a:0.1%v/v tfa
·
h2o;
[0158]
流动相b:0.1%v/v tfa
·
ch3cn;
[0159]
洗脱条件:最初用8%v/v的流动相b进行色谱柱的平衡,随后在90min以内用8%-80%v/v的流动相b进行梯度洗脱,然后使用维持在80%v/v的流动相b进行等度洗脱,最后使用降至8%v/v的流动相b进行色谱柱的再平衡。
[0160]
方法b:rp-hplc
[0161]
色谱柱填料:c18 smb200-10;
[0162]
检测波长:230nm;
[0163]
流动相a:35mmol/l kh2po4·
h2o,用h3po4调节至ph=3.0
±
0.2;
[0164]
流动相b:ch3cn;
[0165]
洗脱条件:最初用5%v/v的流动相b进行色谱柱的平衡,随后在70min以内用5%-65%v/v的流动相b进行梯度洗脱,然后使用维持在65%v/v的流动相b进行等度洗脱,最后使用降至5%v/v的流动相b进行色谱柱的再平衡。
[0166]
方法c:rp-hplc
[0167]
色谱柱填料:c18 smb200-10;
[0168]
检测波长:230nm;
[0169]
流动相a:1%v/v hac
·
h2o;
[0170]
流动相b:ch3cn;
[0171]
洗脱条件:最初用5%v/v的流动相b进行色谱柱的平衡,随后在90min以内用5%-90%v/v的流动相b进行梯度洗脱,然后使用维持在90%v/v的流动相b进行等度洗脱,最后使用降至5%v/v的流动相b进行色谱柱的再平衡。
[0172]
方法d:rp-hplc
[0173]
色谱柱填料:c18 smb200-10;
[0174]
检测波长:230nm;
[0175]
流动相a:0.02%v/v tfa
·
h2o;
[0176]
流动相b:0.02%v/v tfa
·
ch3cn;
[0177]
洗脱条件:最初用5%v/v的流动相b进行色谱柱的平衡,随后在75min以内用5%-80%v/v的流动相b进行梯度洗脱,然后使用维持在80%v/v的流动相b进行等度洗脱,最后使用降至5%v/v的流动相b进行色谱柱的再平衡。
[0178]
各批次、各纯化工艺的总体检测结果如下:
[0179][0180]
由以上检测结果可知,在纯化工艺(3)的条件下,三个批次产品的产量和纯度均较
高。
[0181]
随后,又检测了各批次、各纯化工艺的产品中所包含的各类成分的含量,检测结果如下:
[0182][0183]
从以上三种不同纯化工艺的样品质量比对结果可以看出,纯化工艺(1)、(2)和(3)的水分含量大致相当,肽含量以纯化工艺(3)为最高,并且纯化工艺(3)的tfa含量最低。反相高效液相色谱法结果表明,采用纯化工艺(3)的3个小批次的样品纯度最高,可达98.0%以上,并且作为已知杂质的cys39脱酰胺和met14氧化杂质、asn28脱酰胺杂质、二聚体杂质含量均处于较低水平,最大未知单杂不超过0.3%;离子色谱法检测结果表明,d-his杂质含量同样较低。另外两种纯化工艺所得的样品在反相高效液相色谱法和离子色谱法条件下检测的纯度明显偏低。
[0184]
实施例三:聚乙二醇化艾塞那肽变体mpeg-ppmal-cys
39-exendin-4的合成
[0185]
在反应瓶中配制ph=6.5的磷酸盐缓冲液,n2置换30-50min,再将艾塞那肽变体cys
39-exendin-4(例如通过实施例二中的纯化方法得到)和mpeg-ppmal(分子量为23
±
2.3kda)按照1:2的摩尔比加入到装有磷酸盐缓冲液的反应瓶中,反应温度控制在22
±
2℃,搅拌反应60min,并取样检测反应进程。反应结束后,反应液用ph=4.0的乙酸钠/乙酸溶液稀释,再滴加适量冰乙酸将混合液的ph值调节至4.0,得到聚乙二醇化艾塞那肽变体mpeg-ppmal-cys
39-exendin-4粗品溶液。
[0186]
实施例四:聚乙二醇化艾塞那肽变体mpeg-ppmal-cys
39-exendin-4的纯化
[0187]
按照实施例三中记载的方法,制备三个批次的聚乙二醇化艾塞那肽变体mpeg-ppmal-cys
39-exendin-4粗品溶液,每一批粗品溶液均分成四份。
[0188]
分别采用下列纯化工艺(一)-(四)对每一批粗品溶液中的四份样品进行纯化,并进行相关检测:
[0189]
纯化工艺(一):方法a
→
方法b
→
方法a
→
方法c;
[0190]
纯化工艺(二):方法d
→
方法c;
[0191]
纯化工艺(三):方法a
→
方法b
→
方法a
→
方法d
→
方法c;
[0192]
纯化工艺(四):方法d
→
方法a
→
方法b
→
方法a
→
方法c。
[0193]
在上述纯化工艺中,纯化工艺(三)和(四)都需要经历四步纯化,区别在于,纯化工
艺(三)先进行三步反相色谱纯化,再进行一步离子交换纯化,而纯化工艺(四)先进行一步离子交换纯化,再进行三步反相色谱纯化。
[0194]
上述纯化工艺中使用的方法a-d的具体情况如下:
[0195]
方法a:rp-hplc
[0196]
色谱柱填料:c18 smb200-10;
[0197]
检测波长:230nm;
[0198]
流动相a:0.1%v/v tfa
·
h2o;
[0199]
流动相b:0.1%v/v tfa
·
ch3cn;
[0200]
洗脱条件:最初用9%v/v的流动相b进行色谱柱的平衡,随后在70min以内用9%-80%v/v的流动相b进行梯度洗脱,然后使用维持在80%v/v的流动相b进行等度洗脱,最后使用降至9%v/v的流动相b进行色谱柱的再平衡。
[0201]
方法b:rp-hplc
[0202]
色谱柱填料:c18 smb200-10;
[0203]
检测波长:230nm;
[0204]
流动相a:35mmol/l kh2po4·
h2o,用h3po4调节至ph=3.0
±
0.2;
[0205]
流动相b:ch3cn;
[0206]
洗脱条件:最初用8%v/v的流动相b进行色谱柱的平衡,随后在70min以内用8%-65%v/v的流动相b进行梯度洗脱,然后使用维持在65%v/v的流动相b进行等度洗脱,最后使用降至8%v/v的流动相b进行色谱柱的再平衡。
[0207]
方法c:超滤
[0208]
将样品超滤4遍后,用纯化水将其稀释约1倍,再用0.1mol/l nahco3将其ph值调节至4.0-5.0,然后再超滤3遍。若因产品浓度过大而使超滤过程的进行速度缓慢,可适当加入纯化水进行稀释,最终产品浓度约为20~40mg/ml。
[0209]
方法d:离子交换色谱法
[0210]
色谱柱填料:macrocap sp;
[0211]
检测波长:230nm;
[0212]
流动相a:20mmol/l naac,用hac调节至ph=4.0;
[0213]
流动相b:20mmol/l naac 1mol/l nacl,用hac调节至ph=4.0;
[0214]
流动相c:流动相a:流动相b=8:2(v/v);
[0215]
流动相d:0.2mol/l naoh;
[0216]
洗脱条件:
[0217]
步骤1:用1-3cv的流动相d冲洗akta系统,直至电导率基本恒定时间大于5min,且基线基本稳定;
[0218]
步骤2:用1-3cv的流动相b冲洗akta系统,直至电导率基本恒定时间大于5min,且基线基本稳定;
[0219]
步骤3:用1-3cv的流动相a冲洗akta系统,直至电导率基本恒定时间大于5min,且基线基本稳定;
[0220]
步骤4:用2倍样品溶液体积的流动相a将样品稀释均匀,上样到akta系统;
[0221]
步骤5:用2-5cv的流动相a冲洗akta系统,直至电导率基本恒定时间大于15min;
[0222]
步骤6:用2-4cv的流动相c冲洗akta系统,收集组分。
[0223]
各批次、各纯化工艺的检测结果如下:
[0224][0225]
从以上数据可以看出,纯化工艺(一)所得产物中均包含部分游离的peg,而纯化工艺(二)所得产物的纯度较低,无法满足要求;无论是纯度还是收率,纯化工艺(四)生产出的聚乙二醇化艾塞那肽变体mpeg-ppmal-cys
39-exendin-4都优于纯化工艺(三);此外,纯化工艺(三)的整体纯化时间比纯化工艺(四)偏长,超滤所花费的时间也更长,因此纯化工艺(四)更能满足生产需求。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
