1.本发明属于天然药物制备领域,具体地说,涉及一种草乌甲素的制备方法。
背景技术:
2.草乌甲素(bulleyaconitine a,分子式c
35h49
no
10
,分子量643.76)又名滇西嘟拉碱甲,为白色结晶或结晶性粉末,最早是从毛莨科乌头属植物滇西嘟拉(aconitum bulleyanum diels)中分离出的一种具有生物活性的生物碱。因其具有较强的镇痛及免疫调节等药理活性,现如今作为一种现代植物药,广泛应用于风湿性及类风湿性关节炎、骨关节炎、腰肌劳损、肩周炎、四肢扭伤等的治疗。草乌甲素具有镇痛时间长,副作用小,疗效明确且对器官无明显损伤等特点。相比于传统的镇痛药,草乌甲素没有阿片类镇痛药的成瘾性及耐受性等副作用,也没有非甾体消炎药类镇痛药的胃肠道损伤、出血及肾毒性等不良反应。
3.目前上市的草乌甲素药物剂型种类丰富,有草乌甲素片剂、胶囊剂、口服溶液剂、注射剂。临床上对草乌甲素新剂型和新适应症的研究也相继报道。随着草乌甲素制剂在临床上的广泛应用,草乌甲素原料药的需求也在不断增加,现如今,草乌甲素原料药主要来源于乌头属类植物的提取分离。因此,草乌甲素的提取纯化工艺水平将直接决定草乌甲素原料药的质量水平。
4.现有技术中,公开号为cn102775349a的专利申请公开了一种草乌甲素的制备方法,采用室温甲醇或乙醇渗滤提取、硅胶柱层析及重结晶精制,得到含量在98-99%的草乌甲素纯品;公开号为cn104326981a的专利申请公开了一种草乌甲素的高效提取分离方法,采用酸性乙醇溶液热提、硅胶柱层析及重结晶精制等步骤,得到收率在43%左右,含量在98-99%的草乌甲素纯品;公开号为cn106008344a的专利申请公开了一种草乌甲素的制备方法,采用乙醇溶液常温浸提,氧化铝填料柱层析,二元洗脱剂洗脱,丙酮中重结晶等步骤,得到含量在98-99%的草乌甲素纯品;公开号为cn102924376a的专利申请公开了一种高纯度草乌甲素的制备方法,采用匀浆法实现粉碎、提取一步完成,采用大孔树脂及离子交换树脂二次层析,进一步重结晶精制,得到收率低于50%,含量低于99%的草乌甲素纯品。
5.以上方法中,提取步骤采用室温冷浸或热浸,导致提取时间长,提取效率低。采用重结晶精制容易导致草乌甲素转移率低且杂质含量偏高等问题。低转移率(50%以下)及偏低的纯度(草乌甲素含量低于99%)对草乌甲素药物的发展带来了不利的影响。
6.有鉴于此特提出本发明。
技术实现要素:
7.本发明要解决的技术问题在于克服现有技术的不足,提供一种草乌甲素的制备方法,尤其是提取纯化方法,可以大大缩短提取及纯化时间,提高了草乌甲素的转移率、降低了杂质的含量,提高了草乌甲素的纯度。
8.为解决上述技术问题,本发明采用技术方案的基本构思是:
9.本发明提供一种草乌甲素的制备方法,包括以下步骤:
10.(1)粉碎和提取:取乌头块根,粉碎成乌头粗粉;在乌头粗粉中加入乙醇溶液,加热回流提取,合并提取液,减压浓缩至无醇味,回收乙醇溶液,得提取浓缩液;
11.(2)酸沉:在提取浓缩液中加入酸,调节ph,静置;酸沉液抽滤或离心,滤渣用酸水洗涤,过滤后合并酸液;
12.(3)萃取:在酸液中加入碱液,调节ph;用有机溶剂萃取,合并有机相,减压浓缩至干,得草乌甲素萃取浸膏;
13.(4)柱层析粗分离:将草乌甲素萃取浸膏溶解,加入硅胶拌样,硅胶柱层析加压洗脱,收集含草乌甲素的洗脱段,减压浓缩,得到草乌甲素粗分离物;
14.(5)柱层析精分离:将草乌甲素粗分离物溶解,加入硅胶拌样,硅胶柱层析加压洗脱,收集仅含草乌甲素的洗脱段,减压浓缩,得到草乌甲素纯品。
15.本发明的草乌甲素的制备方法,采用醇溶液进行热回流提取的方式代替冷浸提取,可以缩短提取时间,提高提取效率。提取后经过酸沉,碱液调节ph,再进行有机萃取,可以提高草乌甲素的转移率。同时,采用柱层析粗分离和柱层析精分离,这种两次加压柱层析的方式代替传统的一次柱层析纯化和重结晶精制的方式,可以进一步提高草乌甲素的转移率,降低制备的产品中的杂质,进一步提高草乌甲素纯品的纯度。
16.进一步的方案,步骤(1)中,所述乙醇溶液的体积浓度为65-85%;
17.优选的,加入的乙醇溶液的体积为乌头粗粉重量的5-8倍。
18.进一步的方案,步骤(2)中,加入酸调节ph至1-3,静置6-10h;
19.优选的,所述酸选自盐酸、硫酸、磷酸、醋酸中的至少一种。
20.进一步的方案,步骤(3)中,加入碱液后调节ph至6-9;
21.优选的,所述碱液选自氨水溶液、氢氧化钠溶液、氢氧化钾溶液中的至少一种。
22.进一步的方案,步骤(3)中,有机溶剂的体积用量为调节ph后溶液体积的2-4倍;
23.优选的,所述有机溶剂选自乙酸乙酯、乙酸丙酯、丙酮、丁酮、二氯甲烷、三氯甲烷中的至少一种。
24.进一步的方案,步骤(4)中,草乌甲素萃取浸膏用乙酸乙酯溶解,加入1-3倍硅胶拌样,制成样品;
25.柱层析硅胶用量为样品质量的3-6倍。
26.进一步的方案,步骤(4)中,采用石油醚:丙酮的体积比为4:1-10:1的混合液作为洗脱剂,二乙胺调节ph至10-13,进行等梯度洗脱;
27.优选的,采用石油醚:丙酮的体积比为5:1的混合液作为洗脱剂,二乙胺调节ph至12-13;
28.优选的,加压层析洗脱时,控制洗脱速度为1.0-8.0bv/h;优选洗脱速度为2.0-4.0bv/h。
29.进一步的方案,步骤(5)中,将草乌甲素粗分离物用适量丙酮溶解,加入1-3倍硅胶拌样。
30.进一步的方案,步骤(5)中,柱层析硅胶用量为草乌甲素粗分离物的30-60倍。
31.进一步的方案,步骤(5)中,采用石油醚:丙酮的体积为8:1-15:1的混合液作为洗脱剂,二乙胺调节ph至10-13,进行等梯度洗脱;
32.优选的,采用石油醚:丙酮的体积为12:1的混合液作为洗脱剂,二乙胺调节ph至12;
33.优选的,加压层析洗脱时,控制洗脱速度为1.0-4.0bv/h,优选洗脱速度为4.0bv/h。
34.进一步的方案,得到含量为99%以上的草乌甲素纯品,草乌甲素的总转移率为70-85%。
35.本发明中,所述的乌头块根为乌头属植物的块根,本发明的制备方法适用于现有技术中可以作为草乌甲素提取来源的任何类型的乌头属植物,例如长喙乌头等等。
36.草乌甲素纯品中草乌甲素的含量采用高效液相色谱法进行检测,检测方法包括:
37.1、色谱条件用十八烷基硅烷键合硅胶为填充剂;以0.2%三乙胺水溶液(用磷酸调节ph值至3.1
±
0.1)-乙腈(60∶40)为流动相;柱温30℃;检测波长为260nm。理论板数按草乌甲素峰计算应不低于3000。
38.2、测定方法取本品约20mg,精密称定,置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀,精密量取5ml,置50ml量瓶中,用流动相稀释至刻度,摇匀,精密量取20μl注入液相色谱仪,记录色谱图;按面积归一化计算,即得。
39.采用上述技术方案后,本发明与现有技术相比具有以下有益效果。
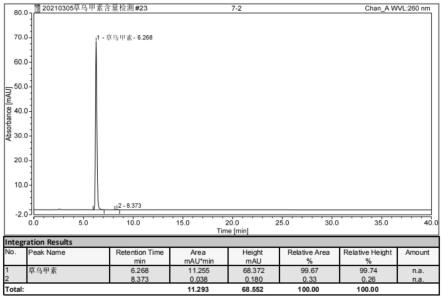
40.(1)本发明采用醇溶液热回流提取代替传统浸泡,可以大大缩短提取时间,提高提取效率。
41.(2)本发明醇溶液热回流提取后,得到的提取浓缩液,先经过酸沉,再碱液调节ph,然后再利用有机溶剂进行萃取,可以提高草乌甲素的转移率。
42.(3)本发明采用二次柱层析分离纯化代替传统一次层析加重结晶,提高了草乌甲素的总转移率,降低了杂质水平,提高了得到的草乌甲素的纯度。
43.(4)本发明的两次柱层析条件不同,分别更适于粗分离和精分离,且采用加压层析代替传统常压层析,缩短了层析时间,提高了层析效率,可以提高分离效果。
44.(5)本发明草乌甲素转移率达到70%以上,纯品草乌甲素含量大于99%,较传统工艺有了明显的提升。
45.下面结合附图对本发明的具体实施方式作进一步详细的描述。
附图说明
46.附图作为本发明的一部分,用来提供对本发明的进一步的理解,本发明的示意性实施例及其说明用于解释本发明,但不构成对本发明的不当限定。显然,下面描述中的附图仅仅是一些实施例,对于本领域普通技术人员来说,在不付出创造性劳动的前提下,还可以根据这些附图获得其他附图。在附图中:
47.图1为实施例1提供的制备方法所得到的草乌甲素的hplc检测结果图;
48.图2为实施例2提供的制备方法所得到的草乌甲素的hplc检测结果图;
49.图3为实施例3提供的制备方法所得到的草乌甲素的hplc检测结果图;
50.图4为对比例1提供的制备方法所得到的草乌甲素的hplc检测结果图;
51.图5为对比例2提供的制备方法所得到的草乌甲素的hplc检测结果图;
52.图6为对比例3提供的制备方法所得到的草乌甲素的hplc检测结果图。
53.需要说明的是,这些附图和文字描述并不旨在以任何方式限制本发明的构思范围,而是通过参考特定实施例为本领域技术人员说明本发明的概念。
具体实施方式
54.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对实施例中的技术方案进行清楚、完整地描述,以下实施例用于说明本发明,但不用来限制本发明的范围。
55.本发明中草乌甲素纯品中草乌甲素的含量采用高效液相色谱法进行检测,检测方法包括:
56.1、色谱条件用十八烷基硅烷键合硅胶为填充剂;以0.2%三乙胺水溶液(用磷酸调节ph值至3.1
±
0.1)-乙腈(60∶40)为流动相;柱温30℃;检测波长为260nm。理论板数按草乌甲素峰计算应不低于3000。
57.2、测定方法取本品约20mg,精密称定,置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀,精密量取5ml,置50ml量瓶中,用流动相稀释至刻度,摇匀,精密量取20μl注入液相色谱仪,记录色谱图;按面积归一化计算,即得。
58.实施例1
59.(1)粉碎和提取:取2kg长喙乌头块根净选后粉碎成粗粉,用75%乙醇溶液回流提取3次,每次1.5小时,乙醇溶液用量为药材质量的7倍(体积重量比)。合并3次提取液,60℃减压浓缩至相对密度为1.20。
60.(2)酸沉:浓缩液冷却至室温后用浓盐酸调节ph至2.0,静置12h,离心过滤,滤饼用适量ph为2.0的盐酸溶液洗涤,合并滤液,得到提取酸液。
61.(3)萃取:用浓氨水调节提取酸液ph为6.5,用4倍体积的乙酸乙酯萃取4次,合并乙酸乙酯层,减压浓缩,得萃取浸膏。
62.(4)柱层析粗分离:用少量乙酸乙酯溶解萃取浸膏,用2倍质量硅胶拌样,6倍质量硅胶粗分离层析。采用石油醚-丙酮(5:1,二乙胺调节ph=12)等度洗脱,加压,控制洗脱速度为2.0bv/h。薄层色谱监测,合并含草乌甲素的洗脱液,减压浓缩至干,得草乌甲素粗分离物。
63.(5)柱层析精分离:用适量丙酮溶解草乌甲素粗分离物,加入3倍质量的硅胶拌样,用40倍质量的硅胶精分离层析。采用石油醚-丙酮(12:1,二乙胺调节ph=12)等度洗脱,加压,控制洗脱速度为4.0bv/h。薄层色谱监测,合并含单一草乌甲素的洗脱液,减压浓缩至干,真空干燥箱内干燥,得草乌甲素纯品4.76g。
64.草乌甲素的转移率为85.0%,经高效液相色谱法检测草乌甲素纯品中草乌甲素含量为99.67%。以上各步骤所述的百分数为质量分数。
65.实施例2
66.(1)粉碎和提取:取2kg长喙乌头块根净选后粉碎成粗粉,用70%乙醇溶液回流提取3次,每次100min,乙醇溶液用量为药材质量的5倍(体积重量比)。合并3次提取液,60℃减压浓缩至相对密度为1.20。
67.(2)酸沉:浓缩液冷却至室温后用浓盐酸调节ph至3.0,静置12h,离心过滤,滤饼用适量ph为3.0的盐酸溶液洗涤,合并滤液,得到提取酸液。
68.(3)萃取:用浓氨水调节提取酸液ph为7.0,用4倍体积的乙酸乙酯萃取4次,合并乙酸乙酯层,减压浓缩,得萃取浸膏。
69.(4)柱层析粗分离:用少量乙酸乙酯溶解萃取浸膏,用2倍质量硅胶拌样,6倍质量硅胶粗分离层析。采用石油醚-丙酮(5:1,二乙胺调节ph=13)等度洗脱,加压,控制洗脱速度为2.0bv/h。薄层色谱监测,合并含草乌甲素的洗脱液,减压浓缩至干,得草乌甲素粗分离物。
70.(5)柱层析精分离:用适量丙酮溶解草乌甲素粗分离物,加入3倍质量的硅胶拌样,用40倍质量的硅胶精分离层析。采用石油醚-丙酮(12:1,二乙胺调节ph=13)等度洗脱,加压,控制洗脱速度为4.0bv/h。薄层色谱监测,合并含单一草乌甲素的洗脱液,减压浓缩至干,得草乌甲素纯品4.14g。
71.草乌甲素转移率为73.93%,经高效液相色谱法检测草乌甲素纯品中草乌甲素含量为99.52%。
72.实施例3
73.(1)粉碎和提取:取2kg长喙乌头块根净选后粉碎成粗粉,用75%乙醇溶液回流提取3次,每次100min,乙醇溶液用量为药材质量的7倍(体积重量比)。合并3次提取液,60℃减压浓缩至相对密度为1.20。
74.(2)酸沉:浓缩液冷却至室温后用浓硫酸调节ph至2.0,静置12h,离心过滤,滤饼用适量ph为2.0的硫酸溶液洗涤,合并滤液,得到提取酸液。
75.(3)萃取:用氢氧化钠固体调节提取酸液ph为7.0,用4倍体积的乙酸乙酯萃取4次,合并乙酸乙酯层,减压浓缩,得萃取浸膏。
76.(4)柱层析粗分离:用少量乙酸乙酯溶解萃取浸膏,用2倍质量硅胶拌样,6倍质量硅胶粗分离层析。采用石油醚-丙酮(5:1,二乙胺调节ph=12)等度洗脱,加压,控制洗脱速度为4.0bv/h。薄层色谱监测,合并含草乌甲素的洗脱液,减压浓缩至干,得草乌甲素粗分离物。
77.(5)柱层析精分离:用适量丙酮溶解草乌甲素粗分离物,加入3倍质量的硅胶拌样,用40倍质量的硅胶精分离层析。采用石油醚-丙酮(12:1,二乙胺调节ph=12)等度洗脱,加压,控制洗脱速度为4.0bv/h。薄层色谱监测,合并含单一草乌甲素的洗脱液,减压浓缩至干,得草乌甲素纯品4.42g。
78.草乌甲素转移率为78.93%,经高效液相色谱法检测草乌甲素纯品中草乌甲素含量为99.60%。
79.对比例1
80.按照专利cn104326981a一种草乌甲素的高效提取分离方法实施例1进行重复试验。草乌甲素的转移率为45.24%,经高效液相色谱法检测草乌甲素纯品中草乌甲素含量为97.56%。
81.对比例2
82.按照专利cn106008344a一种草乌甲素的制备方法实施例1进行重复试验。草乌甲素的转移率为43.26%,经高效液相色谱法检测草乌甲素纯品中草乌甲素含量为98.78%。
83.对比例3
84.按照专利cn101555227a一种高纯度草乌甲素的制备方法实施例2进行重复试验,
草乌甲素的转移率为56.76%,经高效液相色谱法检测草乌甲素纯品中草乌甲素含量为99.58%。
85.实施例及对比例中试验结果如下表1所示:
86.表1
[0087] 草乌甲素转移率(%)草乌甲素含量(%)实施例185.0099.67实施例273.9399.52实施例378.9399.60对比例145.2497.98对比例243.2698.78对比例356.7699.58
[0088]
从表1的试验结果对比可知,使用本发明的方法制备草乌甲素,转移率较现行工艺有了大幅提升,且纯品中草乌甲素含量也能保持较高水平。
[0089]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。