大环类化合物及其中间体的制备方法
发明领域
1.本发明属于药物化学领域,具体涉及一种作为治疗丙型肝炎病毒感染的大环类化合物的制备方法及其重要中间体。
2.发明背景
3.国际申请pct/us2009/056937(wo2011034518)公开了化合物81(即本发明式(i)所示的化合物)及其制备方法,所述化合物对ns3/4a蛋白酶具有很好的抑制活性,并能降低hcv rna水平,可以用于丙型肝炎病毒感染。其中,化合物81的制备方法的合成路线如下所示:
[0004][0005]
其中,该合成路线的步骤2、步骤4和步骤5都需要经过硅胶柱层析纯化才能得到产物,不适合工业化生产。
技术实现要素:
[0006]
针对现有技术中制备式(i)所示化合物存在的问题,本发明对式(i)所示化合物的合成路线进行了优化,提供了一种适合于工业化生产的制备方法。下列反应方案描述了本发明制备如式(i)所示的化合物的合成方法:
[0007][0008]
化合物(vii)脱去boc保护基,得到化合物(vi);化合物(vi)和化合物(v)在nmm和hatu的存在下反应,得到化合物(iv);化合物(iv)脱去boc保护基,得到化合物(iii);化合物(iii)和化合物(iii-1)或其二环己胺盐在nmm和hatu的存在下反应,得到化合物(ii);化合物(ii)在hoveyda-grubbs第二代催化剂的条件下发生烯烃复分解反应得到化合物(i)(含有正庚烷溶残约9000ppm)。
[0009]
另外,本发明还公开了式(i)所示的化合物除去溶残的以下方法:将含有溶残的式(i)所示的化合物溶解于合适的溶剂中(如丙酮等),然后减压蒸馏,残留物用合适溶剂溶解,最后加入到水中析晶。
[0010]
本发明的该制备方法合成步骤较少、操作简单,中间体收率和总产量收率都很高,并且后处理能有效去除杂质和溶残;并且本发明的该制备过程无需使用硅胶柱层析纯化,后处理简便,纯化容易,对设备的要求低,大大降低了生产成本,且过程更加安全可控,适于工业化生产。
[0011]
具体地,一方面,本发明涉及式(i)所示化合物的制备方法,包含以下步骤:以甲苯作为溶剂,式(ii)所示化合物在hoveyda-grubbs第二代催化剂存在的条件下,发生烯烃复分解反应,然后反应液浓缩,所得浓缩残留物经后处理纯化,得到式(i)所示的化合物:
[0012][0013]
在一些实施例中,本发明所述后处理为:
[0014]
步骤1):将浓缩残留物稀释在溶剂1中,加入2-巯基烟酸和三乙胺,在合适温度下搅拌,然后用弱碱性水溶液1洗涤,分层,保留有机相1;
[0015]
步骤2):向步骤1)中的有机相1中加入无机碱1溶液,发生成盐反应,过滤,然后用酸酸化,分层,保留有机相2;
[0016]
步骤3):向步骤2)中的有机相2中加入无机碱2溶液,发生成盐反应,过滤,然后用酸酸化,分层,保留有机相3;
[0017]
步骤4):将步骤3)中的有机相3过滤、滤液减压浓缩,得到浓缩残留物1;步骤5):将步骤4)中的浓缩残留物1稀释在溶剂2中,加入2-巯基烟酸和三乙胺,在合适温度下搅拌,然后用弱碱性水溶液2洗涤,分层,保留有机相4;
[0018]
步骤6)将步骤5)中的有机相4酸化,最后经浓缩和打浆,得到式(i)所示的化合物。
[0019]
在一些实施例中,本发明所述的步骤4)为:将步骤3)中的有机相3经柱层析硅胶过滤、滤液减压浓缩,得到浓缩残留物1;
[0020]
在一些实施例中,本发明所述的步骤4)中的有机相3是经硅胶漏斗过滤的。
[0021]
在一些实施例中,本发明所述的步骤4)的减压浓缩操作是控制在温度≤55℃下操作的。
[0022]
在一些实施例中,本发明所述的步骤1)中的合适温度为55-85℃,优选为65-75℃,更优选为70℃。
[0023]
在一些实施例中,本发明所述的步骤1)中搅拌的搅拌时间为2-6小时,优选为3-5小时,更优选为4小时。
[0024]
在一些实施例中,本发明所述的步骤5)中的合适温度为55-85℃,优选为65-75℃,更优选为70℃。
[0025]
在一些实施例中,本发明所述的步骤5)中搅拌的搅拌时间为2-6小时,优选为3-5小时,更优选为4小时。
[0026]
在一些实施例中,本发明所述的步骤1)中的溶剂1为乙酸异丙酯和正庚烷的混合溶剂。
[0027]
在一些实施例中,本发明所述的步骤1)中的弱碱性水溶液1为碳酸氢钠的水溶液。
[0028]
在一些实施例中,本发明所述的步骤5)中的溶剂2为乙酸异丙酯和正庚烷的混合溶剂;
[0029]
在一些实施例中,本发明所述的步骤5)中的弱碱性水溶液2为碳酸氢钠的水溶液。
[0030]
在一些实施例中,本发明所述的步骤2)的成盐反应是在5-25℃下进行,优选在10-25℃下进行,更优选在10℃下进行。
[0031]
在一些实施例中,本发明所述的步骤3)的成盐反应是在10-30℃下进行,优选在10-25℃下进行,更优选在25℃下进行。
[0032]
在一些实施例中,本发明所述的步骤2)的酸化是在15-30℃下进行,优选在20-30℃下进行,更优选在25℃下进行。
[0033]
在一些实施例中,本发明所述的步骤3)的酸化是在15-30℃下进行,优选在20-30℃下进行,更优选在25℃下进行。
[0034]
在一些实施例中,本发明所述的步骤2)中的酸为盐酸。
[0035]
在一些实施例中,本发明所述的步骤3)中的酸为盐酸。
[0036]
在一些实施例中,本发明所述的步骤2)的无机碱1与式(ii)所示化合物的质量比为0.3:1-0.5:1,优选为0.3:1-0.4:1,更优选为0.35:1。
[0037]
在一些实施例中,本发明所述的步骤2)的无机碱1与式(ii)所示化合物的摩尔比为0.82:1-1.37:1,优选为0.82:1-1.10:1,更优选为0.96:1。在一些实施例中,本发明所述的步骤3)的无机碱2与式(ii)所示化合物的质量比为0.3:1-0.5:1,优选为0.3:1-0.4:1,更优选为0.35:1。
[0038]
在一些实施例中,本发明所述的步骤3)的无机碱2与式(ii)所示化合物的摩尔比为0.82:1-1.37:1,优选为0.82:1-1.10:1,更优选为0.96:1。
[0039]
在一些实施例中,本发明所述的步骤2)的无机碱1溶液为氢氧化钠水溶液、氢氧化钾水溶液、氢氧化锂水溶液、氢氧化钙水溶液或氢氧化镁水溶液;
[0040]
在一些实施例中,本发明所述的步骤3)的无机碱2溶液为氢氧化钠水溶液、氢氧化钾水溶液、氢氧化锂水溶液、氢氧化钙水溶液或氢氧化镁水溶液。
[0041]
在一些实施例中,本发明所述烯烃复分解反应的反应温度为60-100℃,优选70-100℃,更优选75-90℃。本发明烯烃复分解反应式(ii)化合物的投料方式,可以一次投料,也可以分多次投料,比如分三次投料、五次投料等;所述的步骤1)中的洗涤,可以根据情况重复多次,比如洗涤2次、3次或4次;步骤3)中过滤所得的滤饼可以根据实际情况选择合适的溶剂淋洗,比如用乙酸异丙酯和正庚烷的混合溶剂淋洗滤饼;所述的步骤5)的洗涤可以根据情况重复多次,比如洗涤2次、3次或4次;式(ii)化合物的烯烃复分解反应的溶剂量和后处理所用溶剂量等,可以根据实际情况做合适的选择。
[0042]
在一些实施例中,本发明所述的式(ii)所示化合物可以由以下制备方法得到:
[0043][0044]
在溶剂a中,式(iii)所示化合物与式(iii-1)所示化合物或其二环己胺盐,在hatu
和n-甲基吗啡啉的存在下,发生缩合反应,得到式(ii)所示化合物。
[0045]
在一些实施例中,本发明所述的溶剂a为丙酮。
[0046]
本发明式(ii)所示化合物制备过程中发生的缩合反应的溶剂量、后处理操作和后处理所用溶剂量等,可以根据实际情况做合适的选择。
[0047]
在一些实施例中,本发明所述的式(iii)所示化合物可以由以下制备方法得到:
[0048][0049]
步骤a)在溶剂a1中,式(v)所示化合物与式(vi)所示化合物在hatu和n-甲基吗啡啉作用下发生缩合反应,得到式(iv)所示的化合物,步骤b)步骤a)得到的式(iv)所示的化合物,在酸性条件a的条件下,继续脱除叔丁氧羰基保护基,得到式(iii)所示的化合物。
[0050]
在一些实施例中,本发明所述的步骤a)的投料方式为:将式(v)所示化合物和hatu加入到溶剂a1中,然后在≤30℃下滴加n-甲基吗啡啉,搅拌一段时间后再加入式(vi)所示化合物。
[0051]
在一些实施例中,本发明所述的步骤a)的投料方式为:将式(v)所示化合物和hatu加入到溶剂a1中,然后在≤30℃下滴加n-甲基吗啡啉,反应混合物搅拌1-2小时后,再加入式(vi)所示化合物。
[0052]
在一些实施例中,本发明所述的步骤a)的反应过程中可适当需要补加溶剂a1。
[0053]
在一些实施例中,本发明所述的溶剂a1为四氢呋喃。
[0054]
在一些实施例中,本发明所述的步骤a)中式(v)所示化合物与式(vi)所示化合物的摩尔比为1.0:1.1-1.0:1.5,优选为1.0:1.1-1.0:1.25,更优选为1.0:1.1。
[0055]
在一些实施例中,本发明所述的步骤b)还包含式(iii)所示化合物的以下纯化方法:
[0056]
步骤1-1)反应结束后的混合体系用甲基叔丁基醚析晶;
[0057]
步骤1-2)步骤1-1)中析出的固体用甲基叔丁基醚冲洗,然后用四氢呋喃和水溶解,再加入水析晶;
[0058]
步骤1-3)步骤1-2)中析出的固体用四氢呋喃和水的混合溶剂淋洗,经水打浆,再烘干,得到纯化的式(iii)所示的化合物。
[0059]
在一些实施例中,本发明所述的酸性条件a为盐酸条件或氯化亚砜和甲醇条件。
[0060]
本发明所述的步骤b)的式(iii)所示化合物纯化所用到的溶剂量,可以根据实际情况做合适的选择。
[0061]
在一些实施例中,本发明所述的式(vi)所示化合物可以由以下制备方法得到:
[0062]
[0063]
式(vii)所示化合物在溶剂b和酸性条件b下,脱除叔丁氧羰基保护基,得到式(vi)所示的化合物。
[0064]
在一些实施例中,本发明所述的溶剂b为四氢呋喃或二氯甲烷;所述酸性条件b为盐酸条件或氯化亚砜和甲醇条件。
[0065]
在一些实施例中,本发明所述的还包括去除式(i)所示的化合物中溶残的方法:
[0066]
步骤(1):将含有溶残的式(i)所示的化合物溶于溶剂a1中,然后减压蒸馏;
[0067]
步骤(2):将步骤(1)得到的蒸馏残留物溶于溶剂a1中,然后加入到水中析晶。
[0068]
在一些实施例中,本发明所述的溶剂a1为丙酮。
[0069]
在一些实施例中,本发明所述的步骤(1)需要重复1、2、3、4、5或6次。
[0070]
另外,需要说明的是,本发明所述的去除式(i)所示化合物溶残,是指将式(i)所示化合物中的乙酸丙酯和正庚烷残留含量降低至合格的标准,当式(i)所示化合物中的乙酸丙酯和正庚烷溶残的总含量小于0.5%为合格。
[0071]
另一方面,本发明还涉及以下其中之一结构的化合物或其盐:
[0072][0073][0074]
本发明所用到的反应溶剂量、一般后处理操作和后处理所用溶剂量等,可以根据实际情况做合适的选择。
[0075]
本发明的详细说明
[0076]
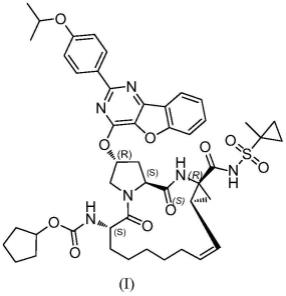
本发明提供了一种作为ns3/4a蛋白酶抑制剂的化合物(2r,6s,13as,14ar,16as,z)-2-(2-(4-异丙氧基苯基)苯并呋喃[3,2-d]嘧啶-4-基氧基)-14a-(1-甲基环丙基磺酰基
氨基甲酰基)-5,16-二氧代-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-十六氢环丙烷[e]吡咯并[1,2-a][1,4]二氮杂环十五烯-6-基氨基甲酸环戊酯(式(i)所示化合物)的制备方法及其重要中间体,本领域技术人员可以借鉴本发明内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明中。
[0077]
定义和一般术语
[0078]
除非另有说明,本发明所用在说明书和权利要求书中的术语具有下述定义。
[0079]
现在详细描述本发明的某些实施方案,其实例由随附的结构式和化学式说明。本发明意图涵盖所有的替代、修改和等同技术方案,它们均包括在如权利要求定义的本发明范围内。本领域技术人员应认识到,许多与本发明所述类似或等同的方法和材料能够用于实践本发明。本发明绝不限于本发明所述的方法和材料。在所结合的文献、专利和类似材料的一篇或多篇与本技术不同或相矛盾的情况下(包括但不限于所定义的术语、术语应用、所描述的技术,等等),以本技术为准。
[0080]
应进一步认识到,本发明的某些特征,为清楚可见,在多个独立的实施方案中进行了描述,但也可以在单个实施例中以组合形式提供。反之,本发明的各种特征,为简洁起见,在单个实施方案中进行了描述,但也可以单独或以任意适合的子组合提供。
[0081]
除非另外说明,本发明所使用的所有科技术语具有与本发明所属领域技术人员的通常理解相同的含义。本发明涉及的所有专利和公开出版物通过引用方式整体并入本发明。
[0082]
除非另外说明,应当应用本发明所使用的下列定义。出于本发明的目的,化学元素与元素周期表cas版,和《化学和物理手册》,第75版,1994一致。此外,有机化学一般原理可参考"organic chemistry",thomas sorrell,university science books,sausalito:1999,和"march's advanced organic chemistry”by michael b.smith and jerry march,john wiley&sons,new york:2007中的描述,其全部内容通过引用并入本发明。
[0083]
除非另有说明或者上下文中有明显的冲突,本发明所使用的冠词“一”、“一个(种)”和“所述”旨在包括“至少一个”或“一个或多个”。因此,本发明所使用的这些冠词是指一个或多于一个(即至少一个)宾语的冠词。例如,“一组分”指一个或多个组分,即可能有多于一个的组分被考虑在所述实施方案的实施方式中采用或使用。
[0084]
本发明所使用的术语“当量”或“eq”数,是按照化学反应的当量关系,以每步中所用基本原料为基准(1当量),所需要的其他原材料的当量用量。
[0085]
本发明“frv03”代表式(ii)所示化合物。
[0086]
术语“包含”为开放式表达,即包括本发明所指明的内容,但并不排除其他方面的内容。
[0087]
术语“室温”是指10℃~40℃,在一些实施方案中,“室温”是指10℃~30℃;还在一些实施方案中,“室温”是指20℃~30℃。
[0088]
另外,需要说明的是,除非以其他方式明确指出,在本发明中所采用的描述方式“各
…
独立地为”与
“…
各自独立地为”和
“…
独立地为”可以互换,均应做广义理解,其既可以是指在不同基团中,相同符号之间所表达的具体选项之间互相不影响,也可以表示在相同的基团中,相同符号之间所表达的具体选项之间互相不影响。
[0089]
术语“保护基团”是指一个取代基与其他官能团起反应的时候,通常用来阻断或保护的特殊功能性。例如,“氨基的保护基团”是指一个取代基与氨基基团相连来阻断或保护化合物中氨基的功能性,合适的氨基保护基团包括乙酰基,三氟乙酰基,叔丁氧羰基(boc,boc),苄氧羰基(cbz,cbz)和9-芴亚甲氧羰基(fmoc)。相似地,“羟基保护基团”是指羟基的取代基用来阻断或保护羟基的功能性,合适的保护基团包括苄基(bn)、苄氧羰基(cbz)、三苯基甲基、对甲氧基苄基(pmb)、叔丁基二甲基硅基(tbdms)、三甲基硅基(tms)、叔丁基二苯基硅基(tbdps)、三乙基硅基(tes)、三异丙基硅基(dips)、2-(三甲硅烷基)乙氧甲基、二氢吡喃基、溴丙烯基、乙酯甲酰基、乙酰基或苯甲酰基。“羧基保护基团”是指羧基的取代基用来阻断或保护羧基的功能性,一般的羧基保护基包括-ch2ch2so2ph,氰基乙基,2-(三甲基硅烷基)乙基,2-(三甲基硅烷基)乙氧基甲基,2-(对甲苯磺酰基)乙基,2-(对硝基苯磺酰基)乙基,2-(二苯基膦基)乙基,硝基乙基,等等。对于保护基团一般的描述可参考文献:t w.greene,protective groups in organic synthesis,john wiley&sons,new york,1991;and p.j.kocienski,protecting groups,thieme,stuttgart,2005.
[0090]
在本发明的上下文中,所有在此公开了的数字均为近似值。每一个数字的数值有可能会出现1%、2%、5%、7%、8%或10%等差异。每当公开一个具有n值的数字时,任何具有n /-1%,n /-2%,n /-3%,n /-5%,n /-7%,n /-8%或n /-10%值以内的数字会被明确地公开,其中“ /
‑”
是指加或减。每当公开一个数值范围中的一个下限,dl,和一个上限,du,时,任何处于该公开了的范围之内的数值会被明确地公开,比如反应于25℃温度下反应,包括反应在25
±
5℃下反应。
[0091]
本发明所述的所有反应步骤反应到一定程度如原料消耗大约大于70%,大于80%,大于90%,大于95%,或经检测反应原料已经消耗完毕后进行后处理,如冷却,收集,提取,过滤,分离,净化处理或其组合。可以通过常规的方法如薄层层析法(tlc)、高效液相色谱法(hplc)、气相色谱法(gc)等方法检测反应程度。可以采用常规的方法对反应溶液进行后处理,例如,通过减压蒸馏或常规蒸馏反应溶剂后收集粗产物,直接投入下一步反应;或直接过滤得到粗产物,直接投入下一步反应;或静置后,倾倒出上层清液得到粗产物,直接投入下一步反应;或选择适当的有机溶剂或其组合进行萃取,蒸馏,结晶,柱层析,润洗,打浆等纯化步骤。
[0092]
本发明所述的各反应步骤所使用的溶剂没有特别限制,任何在一定程度上能溶解起始原料并且不抑制反应的溶剂均包含在本发明中。另外,本领域的许多类似改动,等同替换,或等同于本发明所描述的溶剂,溶剂组合,及溶剂组合的不同比例,均视为本发明的包含范围。本发明给出了各反应步骤所使用的较佳的溶剂。
[0093]
本发明所述的溶剂中水分的含量,没有特别的限制,即,溶剂中水分的含量不影响本发明所述反应的发生。任何在一定程度上能在本发明中使用的含有一定量的水分的溶剂,均视为本发明所述的溶剂。如溶剂中水分的含量大约小于0.05%,小于0.1%,小于0.2%,小于0.5%,小于5%,小于10%,小于25%,小于30%,或为0%。在一些实施方案中,所述溶剂的水分含量在一定范围内,更有利于反应的进行;例如,在以乙醇作为反应溶剂的步骤,使用无水乙醇,更有利反应的进行。在一些实施方案中,所述溶剂的水分含量超出一定范围,可能会影响反应的进行(例如,影响反应的收率),但并不影响反应的发生。
[0094]
一般合成和检测方法
[0095]
在本说明书中,如果在化学名称和化学结构间存在任何差异,以结构为准。
[0096]
所属领域的技术人员将认识到:本发明所描述的化学反应可以用来合适地制备许多与本发明所述的化合物相似的化合物。所属领域的技术人员通过修饰方法,如适当的保护基团,通过利用其他已知的试剂除了本发明所描述的,或将反应条件做一些常规的修改也可以实现本发明,这些常规的制备方法修改也应当认为是属于本发明的范围。另外,本发明所公开的反应或已知的反应条件也公认地适用于其他与本发明所述的化合物相似的化合物的制备。
[0097]
一般地,本发明所描述的方法能制备得到本发明如式(i)所示的化合物。下面的实施例用于进一步举例说明本发明的内容。
[0098]
化合物的结构是通过核磁共振(1h-nmr、
13
c-nmr)来确定的。1h-nmr、
13
c-nmr化学位移(δ)以百万分之一(ppm)的单位给出。1h-nmr、
13
c-nmr的测定是用bruker ultrashield-400核磁共振谱仪和bruker avance iii hd 600核磁共振谱仪,测定溶剂为氘代氯仿(cdcl3)、氘代甲醇(cd3od)或者氘代dmso(dmso-d6),用tms(0ppm)或氘代氯仿(7.26ppm)作为参照标准。当出现多重峰的时候,将使用下面的缩写:s(singlet,单峰),d(doublet,双峰),t(triplet,三重峰),m(multiplet,多重峰),br(broadened,宽峰),dd(doublet of doublets,四重峰),dt(doublet of triplets,双三重峰),ddd(doublet of doubletof doublets,双双二重峰),ddt(doublet of doublet of triplets,双双三重峰),td(triplet of doublets,三双重峰),brs(broadened singlet,宽单峰)。偶合常数j,用赫兹(hz)表示。氢谱中的overlap表示重叠峰。
[0099]
溶剂残留检测用:agilent 7890a-7697气相色谱仪;agilent db-1(60m
×
0.53mm
×
3.0μm)色谱柱。
[0100]
lc-ms的测定用agilent 1200hplc;agilent 6320离子阱质谱仪(tg-0502535-a);agilent 1260hplc;agilent 6530q-tof高分辨质谱仪(tg-2349、tg-2349-a、tg-2349-b、tg-2349-c);
[0101]
本发明的起始原料是已知的,并且可以在市场上购买到得,购买自爱思特(成都)生物制药股份有限公司(astatech(chengdu)biopharmaceutical corp.)、海门慧聚药业有限公司(wisdom pharmaceutical co.,ltd)等公司,或者按照本领域已知的方法来合成。
[0102]
实施例中无特殊说明的,反应均在氮气氛下进行;
[0103]
氮气氛是指反应瓶连接一个约1l容积的氮气气球或钢釜;
[0104]
实施例中无特殊说明,溶液是指水溶液。
[0105]
实施例中无特殊说明,反应温度为室温;
[0106]
本发明杂质含量采用的是hplc测试,hplc是指高效液相色谱;
[0107]
hplc的测定使用安捷伦1200高压液相色谱仪(zorbax eclipse plus c18 150
×
4.6mm色谱柱);
[0108]
hplc测试条件:运行时间:52min柱温:25℃pda:254nm
[0109]
流动相:a相:1ml tfa加入到2000ml纯化水 b相:1ml tfa加入到2000ml乙腈 流速:1.0ml/min
[0110]
下面简写词的使用贯穿本发明:
[0111]
mtbe
ꢀꢀꢀꢀꢀꢀꢀ
甲基叔丁基醚
ꢀꢀꢀꢀꢀꢀꢀ
nmm
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
n-甲基吗啉
[0112]
thf
ꢀꢀꢀꢀꢀꢀꢀꢀ
四氢呋喃
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
dcm
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二氯甲烷
[0113]
ea
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
乙酸乙酯
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
dmso
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二甲基亚砜
[0114]
mass%
ꢀꢀꢀꢀꢀ
质量百分比
ꢀꢀꢀꢀꢀꢀꢀꢀ
hoat
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1-羟基-7-氮杂苯并三氮唑
[0115]
licl
ꢀꢀꢀꢀꢀꢀꢀ
氯化锂
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
min
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
分钟
[0116]hꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
小时
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
r.t.
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
室温
[0117]
meoh
ꢀꢀꢀꢀꢀꢀꢀ
甲醇
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
etoh
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
乙醇
[0118]
hatu 2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯
[0119]
hoveyda-grubbs 2nd generation catalyst hoveyda-grubbs第二代催化剂
[0120]
hoveyda-grubbs第一代催化剂(cas:2037147-71-0);
[0121]
grubbs第二代催化剂(cas:246047-72-3);
[0122]
hoveyda-grubbs第二代催化剂(cas:301224-40-8);
[0123]
zhan 1b催化剂(cas:918870-76-75)。
具体实施方式
[0124]
本发明实施例公开了制备(2r,6s,13as,14ar,16as,z)-2-(2-(4-异丙氧基苯基)苯并呋喃[3,2-d]嘧啶-4-基氧基)-14a-(1-甲基环丙基磺酰基氨基甲酰基)-5,16-二氧代-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-十六氢环丙烷[e]吡咯并[1,2-a][1,4]二氮杂环十五烯-6-基氨基甲酸环戊酯(式(i)所示化合物)及其中间体的方法。
[0125]
本领域技术人员可以借鉴本发明内容,或者适当改进工艺参数来实现本发明的内容。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明的范围中。本发明的方法已经通过实施例进行了描述,相关人员明显能在不脱离本发明内容和范围内对本发明所述的方法进行改动或适当变更与组合,来实现和应用本发明的技术。
[0126]
为了进一步理解本发明,下面结合实施例对本发明进行详细说明。
[0127]
实施例
[0128]
实施例1:(1r,2s)-1-氨基-n-((1-甲基环丙基)磺酰基)-2-乙烯基环丙烷-1-甲酰
胺盐酸盐(式(vi)所示化合物)的制备:
[0129][0130]
于1000ml四口瓶中,依次加式(vii)所示化合物(20g,1.0eq)和四氢呋喃(300ml),最后加入甲醇(14.2ml,6.0eq),室温条件下缓慢滴加氯化亚砜(12.2ml,3.0eq),控制内温不超过40℃,滴毕,转移至45℃反应8小时,降温至室温,向反应体系中加入甲基叔丁基醚(300ml),室温搅拌2小时,抽滤,滤饼用甲基叔丁基醚洗涤(20ml)淋洗,得到固体,将固体加入到四氢呋喃(200ml)中室温打浆12小时,抽滤,滤饼用四氢呋喃(10ml)洗涤,45℃真空干燥12h,得到类白色固体式(vi)所示化合物(16g,产率98.13%,纯度:99.70%)。
[0131]
ms(esi,pos.ion)m/z:245[m h]
;
[0132]1h nmr(600mhz,dmso-d6)δ(ppm)9.16(3h,brs),5.54(1h,m),5.35(1h,d,j=17.0hz),5.21(1h,d,j=10.6hz),2.38(1h,m),2.01(1h,t,j=7.1hz),1.70(1h,dd,j=9.8,6.6hz),1.46(1h,overlap),1.44(3h,s),1.35(1h,m),0.93(2h,m);
[0133]
13
c nmr(151mhz,dmso-d6)δ(ppm)165.2,131.4,119.5,41.6,36.2,28.7,17.4,15.9,13.2,12.2.
[0134]
实施例2:(2s,4r)-4-((2-(4-异丙氧基苯基)苯并呋喃[3,2-d]嘧啶-4-基)氧基)-n-((1r,2s)-1-((1-甲基环丙基)磺酰基)氨甲酰基)-2-乙烯基环丙基)吡咯烷-2-甲酰胺盐酸盐(式(iii)所示化合物)的制备:
[0135][0136]
步骤a)(2s,4r)-4-((2-(4-异丙氧苯基)苯并呋喃[3,2-d]嘧啶-4-基)氧)-2-(((1r,2s)-1-(((1-甲基环丙基)磺酰基)胺甲酰基)-2-乙烯基环丙基)胺甲酰基)吡咯烷-1-羧酸叔丁酯(式(iv)所示化合物)的合成:
[0137]
方法一:取式(v)所示化合物(10g,1.0eq)、hatu(10.7g,1.5eq)和式(vi)所示化合物(6.6g,1.1eq)加入到thf(120ml)中,于25
±
5℃搅拌5min。滴加nmm(5.74g,3.0eq),滴加过程中内温不超过30℃。滴加完毕后于25℃反应3h。将上述反应液过滤,用thf(30ml)淋洗滤饼,滤液直接用于后续式(iii)所示化合物的制备。为了鉴定式(iv)所示化合物,取了部分少量滤液浓缩,经硅胶柱纯化处理。
[0138]
ms(esi,pos.ion)m/z:760.2986[m h]
;1hnmr(600mhz,cdcl3)δ(ppm)9.93(1h,s),8.42(2h,d,j=8.3hz),8.23(1h,d,j=7.5hz),7.63(2h,overlap),7.56(1h,s),7.46(1h,m),7.00(2h,d,j=8.3hz),6.03(1h,s),5.77(1h,m),5.28(1h,d,j=17.0hz),5.14(1h,d,j=10.3hz),4.67(1h,m),4.45(1h,t,j=7.5hz),3.98(1h,dd,j=12.4,3.8hz),3.92(1h,d,
j=12.5hz),2.65(1h,m),2.62(1h,m),2.16(1h,m),1.99(1h,m),1.73(1h,m),1.62(1h,m),1.52(3h,s),1.49(9h,s),1.43(1h,m),1.39(6h,d,j=5.6hz),0.88(1h,m),0.83(1h,m);
[0139]
13
c nmr(151mhz,cdcl3)δ(ppm)173.4,167.8,160.0,159.2,157.6,155.3,153.2,151.3,133.9,132.8,130.8,129.8,129.8,129.8,124.0,122.5,122.1,118.5,115.6,115.6,112.7,81.9,75.2,69.9,59.2,53.0,41.8,36.7,35.4,35.2,28.4,28.4,28.4,22.9,22.1,22.1,18.3,14.2,13.2.
[0140]
参考方法一的合成方法,考察不同反应溶剂对反应的影响,实验结果见表1.
[0141]
表1反应溶剂的影响
[0142][0143][0144]
表1结果表明,thf为反应溶剂,反应状态好,易于搅拌,操作简单,所得反应液无需进一步处理即可用于下一步反应,且对下一步反应的后处理基本无影响,因而可以缩短操作时间并减少生产成本。
[0145]
方法二:将式(v)所示化合物(30.0kg,1.0eq)和hatu(32.0kg,1.5eq)加入到四氢呋喃(310.0kg,10.33g/1g式(v)所示化合物)中,滴加n-甲基吗啡啉(17.0kg,3.0eq),滴加过程中体系内温度不超过30℃。滴加完毕后于25
±
5℃搅拌1h,然后往该体系中加入式(vi)所示化合物(17.4kg,1.1eq),于20~30℃保温反应3h。将上述反应液过滤,用thf(84.0kg,2.8g/1g式(v)所示化合物)淋洗滤饼,滤液直接用于后续式(iii)所示化合物的制备。ms(esi,pos.ion)m/z:760.2986[m h]
。
[0146]
当反应溶剂为thf,参考方法一或方法二的合成方法,考察投料量比例和投料顺序的改变对反应的影响,实验结果如表2所示。
[0147]
表2:投料量比例和投料顺序发生改变后的实验结果
[0148][0149]
表2数据表明,相对于方法一的投料方式,按照方法二的投料顺序投料,反应中杂质含量明显降低,产率也有较大提高;并且当式(v)所示化合物:式(vi)所示化合物的摩尔
量为1.0:1.1时,反应中胺化杂质进一步降低,产率也进一步的提高。
[0150]
步骤b)(2s,4r)-4-((2-(4-异丙氧基苯基)苯并呋喃[3,2-d]嘧啶-4-基)氧基)-n-((1r,2s)-1-((1-甲基环丙基)磺酰基)氨甲酰基)-2-乙烯基环丙基)吡咯烷-2-甲酰胺盐酸盐(式(iii)所示化合物)的合成:
[0151]
直接将步骤a)方法二的滤液加入反应釜中,然后加入四氢呋喃(528.0kg,20ml/1g式(v)所示化合物)和甲醇(28.5kg,15.0mol/1mol式所示(v)化合物)。向反应釜中缓慢滴加的二氯亚砜(51.0kg,7.5mol/1mol式所示(v)化合物),控制内温不超过40℃。滴加完毕后,控制釜内温度45℃反应8h,反应结束。
[0152]
后处理方法一:将上述步骤b)反应完得到的反应体系降温至25℃,往釜内加入甲基叔丁基醚(20ml/1g式(v)所示化合物),保温搅拌2h,然后过滤,滤饼用甲基叔丁基醚(2ml/1g式(v)所示化合物)淋洗,所得滤饼加入四氢呋喃(6.8ml/1g式(v)所示化合物),并于25℃搅拌0.5h,然后再加入水(2ml/1g式(v)所示化合物),搅拌至体系溶清。搅拌下,然后将所得溶液加入到水(1g式(v)所示化合物/34ml)中,于25
±
5℃打浆3h。将上述体系过滤,滤饼用四氢呋喃(0.6ml/1g式(v)所示化合物)/水(3ml/1g式(v)所示化合物)混合溶剂淋洗,所得滤饼加入水(10ml/1g式(v)所示化合物)中,于25
±
5℃打浆2h。将上述体系直接放料、离心,滤饼用水(1ml/1g式(v)所示化合物)淋洗,出料。60
±
5℃真空干燥52h后取样检测,至水分≤5.0%,结束干燥,得到式(iii)所示化合物:36.30kg,收率:92.7%(两步产率)。
[0153]
后处理方法二:按照上述步骤a)方法二和步骤b)的反应操作,将得到的反应体系降温至25℃,往釜内加入的甲基叔丁基醚(20ml/1g式(v)所示化合物),保温搅拌2h。体系直接放料、分离,滤饼用四氢呋喃(0.3ml/1g式(v)所示化合物)/水(1.5ml/1g式(v)所示化合物)混合溶剂淋洗。将所得滤饼于50℃减压烘料,32h结束干燥,得到式(iii)所示化合物。
[0154]
后处理方法三:按照上述步骤a)方法二和步骤b)的反应操作,将得到的反应体系降温至25℃,反应液中加入水(130ml/1g式(v)所示化合物),室温下搅拌3h。抽滤,滤饼用水(1ml/1g式(v)所示化合物)洗涤,滤饼于45℃鼓风干燥15h,得到式(iii)所示化合物。
[0155]
ms(esi,pos.ion)m/z:660.2496[m h]
;
[0156]1h nmr(600mhz,cdcl3)δ(ppm)10.87(1h,brs),10.02(1h,brs),9.18(1h,s),8.68(1h,brs),8.14(2h,d,j=8.0hz),7.92(1h,d,j=7.3hz),7.46(1h,d,j=7.8hz),7.38(1h,t,j=7.2hz),7.16(1h,t,j=7.2hz),6.86(2h,d,j=8.3hz),5.87(1h,s),5.54(1h,m),5.26(1h,d,j=17.1hz),5.17(1h,s),5.05(1h,d,j=10.4hz),4.61(1h,m),4.19(1h,s),4.00(1h,d,j=12.1hz),2.97(1h,s),2.66(1h,s),2.34(1h,m),2.04(1h,m),1.64(1h,m),1.45(1h,m),1.41(3h,s),1.38(6h,d,j=5.9hz),1.35(1h,m),0.68(2h,m);
[0157]
13
c nmr(151mhz,cdcl3)δ(ppm)170.2,167.8,159.9,158.6,157.3,152.3,150.8,133.2,132.2,130.8,129.8,129.8,129.1,123.8,122.0,121.7,118.9,115.4,115.4,112.5,74.7,69.9,59.6,51.6,42.7,36.6,35.7,33.0,22.1,22.0,20.5,18.2,14.5,12.6.
[0158]
参考步骤b)的合成方法,考察采用不同的后处理对实验结果的影响,实验结果见表3。
[0159]
表3:不同后处理的实验结果。
[0160][0161]
表3实验结果表明,方法一和方法三的后处理方法,产物纯度高,杂质含量少,但方法三的析晶过程耗水量过大,不利于工业扩大生产。
[0162]
实施例3:((s)-1-((2s,4r)-4-((2-(4-异丙氧基苯基)苯并呋喃[3,2-d]嘧啶-4-基)氧基)-2-(((1r,2s)-1-(((1-甲基环丙基)磺酰基)氨基甲酰基)-2-乙烯基环丙基)氨基甲酰基)吡咯烷-1-基)-1-酮-8-烯-2-基)氨基甲酸环戊酯(式(ii)所示化合物)的制备:
[0163][0164]
合成方法:向反应釜中先加入式(iii)所示化合物(36.15kg,1.0eq)、式(iii-1)所示化合物的二环己胺盐(26.6kg,1.2eq)和hatu(31.0kg,1.5eq),然后加入丙酮(180.0kg)。向反应釜中缓慢滴加的n-甲基吗啡啉(17.0kg,3.0eq),控制内温不超过30℃。滴加完毕后,控制釜内温度到45℃开始计时,控温反应3h,反应结束。
[0165]
后处理:1)将上述反应液直接加入到氢氧化钠(9.0kg)/水(680kg)溶液中,控制内温25
±
5℃,继续保温搅拌3h,析出固体。将该体系直接离心,用丙酮(18.0kg)/水(68.0kg)混合溶剂淋洗滤饼,得到式(ii)所示化合物的钠盐。2)搅拌下,将盐酸(24.0kg)/水(290.0kg)溶液缓慢加入到式(ii)所示化合物的钠盐和乙酸异丙酯(640.0kg)的混合物中,然后于25
±
5℃下搅拌1h,然后过滤,滤液静置0.5h后,分离除去下层水相。3)向有机相中缓慢加入氢氧化钠(9.0kg)/水(360kg)溶液,然后于25
±
5℃搅拌3h、离心,用乙酸异丙酯(64.0kg)淋洗滤饼。4)搅拌下,将盐酸(24.0kg)/水(290.0kg)加入到滤饼的乙酸异丙酯(640.0kg)溶液中,并于25
±
5℃继续搅拌1h,然后静置0.5h后分液,分离除去水相。5)浓缩上层有机相,控制t≤55℃减压浓缩至无明显馏分。往所得浓缩残留物中加入正庚烷(120.0kg),再控制t≤55℃减压浓缩至无明显馏分,该操作共重复四次。于25
±
5℃下,所得浓缩残留物用正庚烷(120.0kg)打浆,搅拌2h,然后过滤,滤饼用正庚烷(17.0kg)淋洗,最后于60
±
5℃,真空度0.085mpa以上条件下真空干燥12h,得到式(ii)所示化合物(41.55kg,收率:86.5%,纯度99.3%)。
[0166]
ms(esi,pos.ion)m/z:925.4169[m h]
;
[0167]1h nmr(600mhz,cdcl3)δ(ppm)10.02(1h,s),8.42(2h,d,j=8.6hz),8.33(1h,d,j=6.5hz),7.62(2h,overlap),7.46(1h,m),7.41(1h,s),6.99(2h,d,j=8.8hz),6.16(1h,
s),5.78(1h,overlap),5.76(1h,overlap),5.58(1h,d,j=8.8hz),5.28(1h,d,j=17.2hz),5.14(1h,d,j=10.8hz),4.97(1h,dd,j=17.2,1.6hz),4.91(1h,d,j=10.1hz),4.85(1h,m),4.66(1h,m),4.61(1h,t,j=8.1hz),4.43(1h,m),4.31(1h,d,j=11.9hz),4.12(1h,dd,j=11.8,3.9hz),2.65(2h,m),2.15(1h,m),2.01(3h,overlap),1.71(2h,overlap),1.70(2h,overlap),1.62(4h,overlap),1.60(2h,overlap),1.48(3h,s),1.43(3h,overlap),1.39(6h,d,j=5.8hz),1.37(2h,overlap),1.36(2h,overlap),1.29(2h,m),0.88(1h,m),0.81(1h,m);
[0168]
13
c nmr(151mhz,cdcl3)δ(ppm)173.3,172.5,167.7,160.2,158.8,157.7,156.4,153.2,150.7,138.8,133.9,132.7,131.0,130.0,130.0,129.1,124.1,122.6,121.9,118.5,115.6,115.6,114.4,112.7,77.8,75.6,70.0,60.2,53.1,52.7,41.7,36.6,35.1,34.2,33.6,32.7,32.6,32.2,28.7,28.6,25.3,23.6,23.6,23.5,22.0,22.0,18.4,14.3,13.3.
[0169]
参考实施例3的合成方法,考察反应溶剂对实验结果的影响,实验结果见表4。
[0170]
表4不同反应溶剂的实验结果
[0171][0172]
实施例4:(2r,6s,13as,14ar,16as,z)-2-(2-(4-异丙氧基苯基)苯并呋喃[3,2-d]嘧啶-4-基氧基)-14a-(1-甲基环丙基磺酰基氨基甲酰基)-5,16-二氧代-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-十六氢环丙烷[e]吡咯并[1,2-a][1,4]二氮杂环十五烯-6-基氨基甲酸环戊酯(式(i)所示化合物)的合成
[0173][0174]
方法一:
[0175]
式(ii)所示化合物分三批反应:往干燥洁净釜内分别加入式(ii)所示化合物(12.47kg,1.0eq)和甲苯(1390kg),将体系用氮气置换。将体系升温至80℃(下口鼓氮气)。将hoveyda-grubbs第二代催化剂(132g,0.015eq)溶于甲苯(110kg),在氮气保护下,控温80℃,分四次加入体系中,每0.5h加一次。加料完毕,体系于80℃保温反应2h,反应完成后,t≤
65℃减压浓缩反应体系,收集浓缩物。重复三次上述反应,合并三次反应的浓缩物。
[0176]
参考方法一的方法,研究催化剂种类、温度或/和溶剂的改变对烯烃复分解反应的影响,实验结果见表5:
[0177]
表5:催化剂种类、温度或/和溶剂的改变对烯烃复分解反应的影响实验结果
[0178][0179][0180]
注明:三种其它杂质推测可能为二聚体的杂质构型;hplc峰面积%为未经纯化,通过hplc的所占峰面积百分比研究反应后溶液中各产物、原料和杂质的含量;nd代表未检测到。
[0181]
表5显示:使用hoveyda-grubbs第一代催化剂产物含量低,而使用grubbs第二代催化剂原料的含量太高,不符合中控质量标准。虽然zhan 1b催化剂可以符合中控质量标准,
且产率也较高,但太昂贵。使用hoveyda-grubbs第二代催化剂且用甲苯做溶剂时,杂质含量低,产率高,尤其在反应温度控制在75-90℃,产率最高,且杂质含量最低,符合中控质量标准。而使用hoveyda-grubbs第二代催化剂,当使用meoh、etoh、ea、dcm、mtbe或丙酮做溶剂时,杂质含量高,且产率低。
[0182]
后处理:
[0183]
第一次除重金属:往上述浓缩后的浓缩物中加入乙酸异丙酯(490kg)、正庚烷(128kg)、2-巯基烟酸(11kg)和三乙胺(6kg),升温至70℃,保温搅拌4h,降温至25℃。使用碳酸氢钠(21kg)/水(470kg)洗涤三次,得有机相。
[0184]
第一次成盐、游离:往上述有机相中加入氢氧化钠(13.2kg)/水(400kg)溶液,控制内温10℃,搅拌4h后过滤分离,用乙酸异丙酯(49kg)和正庚烷(13kg)混合液淋洗滤饼。将滤饼和乙酸异丙酯(490kg)和正庚烷(128kg)的混合物液加入反应釜。然后向反应釜中缓慢加入浓盐酸(43kg)/水(210kg),控制内温25℃,搅拌1h,静置0.5h后分液,得上层有机相。
[0185]
第二次成盐、游离:往该有机相中加入氢氧化钠(13.2kg)/水(400kg)溶液,控制内温25℃,搅拌4h后分离,用乙酸异丙酯(49kg)/正庚烷(13kg)的混合物液淋洗滤饼,将滤饼和乙酸异丙酯(490kg)和正庚烷(128kg)的混合物液加入反应釜。向反应釜中缓慢加入浓盐酸(43kg)/水(210kg)溶液,控制内温25℃,搅拌1h,静置0.5h后分液得有机相。
[0186]
过快速硅胶漏斗:铺好滤布,加入柱层析硅胶(42kg),放料过滤,控温t≤55℃,将所得滤液减压浓缩,收集馏分;馏分再淋洗硅胶,控温t≤55℃,将所得滤液减压浓缩,收集馏分。将收集的馏分直接淋洗硅胶,控温t≤55℃,将所得滤液减压浓缩至无明显馏分,得浓缩残留物。
[0187]
第二次除重金属:往上述浓缩残留物中加入乙酸异丙酯(490kg)、2-巯基烟酸(11kg)和三乙胺(6kg),升温至70℃,保温搅拌4h,降温至25℃。向体系中加入碳酸氢钠(21kg)/水(470kg)溶液,搅拌1h,静置0.5h后分液,弃去下层水相,该操作共重复三次。
[0188]
游离、浓缩、打浆、抽滤:控温25℃,然后向上述分离液中缓慢加入浓盐酸(43kg)/水(210kg)溶液,搅拌1h。静置0.5h后弃去下层水相,将有机相在t≤55℃减压浓缩至无明显馏分。向体系中加入正庚烷(120kg),减压浓缩至无明显馏分。降温至25℃。向上述残留中加入正庚烷(120kg),常温打浆搅拌2h后直接抽滤,用正庚烷(12kg)淋洗滤饼,得到滤饼。控制温度60℃真空干燥12h,得到类白色固体为式(i)所示化合物(26.85kg,产率74%,纯度:99.8%,式(v)所示化合物含量:未检出。非特定单杂:0.05%;总杂:0.05%)。
[0189]
ms(esi,pos.ion)m/z:897.3824[m h]
;
[0190]1h nmr(600mhz,dmso-d6)δ(ppm)10.96(1h,s),9.15(1h,s),8.44(2h,d,j=8.8hz),8.22(1h,d,j=7.7hz),7.87(1h,d,j=8.4hz),7.75(1h,t,j=7.8hz),7.54(1h,t,j=7.5hz),7.27(1h,d,j=6.7hz),7.09(2h,d,j=8.9hz),6.15(1h,s),5.62(1h,m),5.03(1h,t,j=9.4hz),4.75(1h,m),4.69(2h,overlap),3.97(1h,m),3.92(2h,overlap),2.66(1h,overlap),2.62(1h,overlap),2.48(1h,m),2.39(1h,m),1.71(1h,overlap),1.69(1h,overlap),1.64(1h,m),1.55(1h,m),1.46(1h,m),1.41(3h,s),1.39(1h,m),1.37(1h,m),1.33(6h,d,j=6.0hz),1.31(2h,m),1.29(1h,overlap),1.26(3h,overlap),1.18(1h,m),1.15(1h,m),1.13(1h,m),1.09(1h,m),1.06(1h,m),1.04(1h,m),0.97(2h,m),0.90(1h,m),0.87(1h,m);
[0191]
13
c nmr(151mhz,dmso-d6)δ(ppm)177.1,172.1,167.7,159.4,158.0,156.9,155.4,153.4,149.9,134.7,134.2,130.9,129.5,129.5,129.3,125.6,124.2,121.8,121.4,115.3,115.3,112.9,76.2,75.6,69.2,59.0,52.3,51.8,43.5,36.0,34.3,31.8,31.4,31.2,29.0,27.0,26.4,26.0,22.8,22.7,21.7,21.7,21.5,21.3,17.3,13.2,11.9.
[0192]
参照实施例4方法一,改变后处理的条件,考察对产率和纯度的影响,实验结果见表6
[0193]
表6:后处理的条件改变对产率和纯度的影响结果
[0194][0195]
实施例5:(2r,6s,13as,14ar,16as,z)-2-(2-(4-异丙氧基苯基)苯并呋喃[3,2-d]嘧啶-4-基氧基)-14a-(1-甲基环丙基磺酰基氨基甲酰基)-5,16-二氧代-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-十六氢环丙烷[e]吡咯并[1,2-a][1,4]二氮杂环十五烯-6-基氨基甲酸环戊酯(式(i)所示化合物)的精制
[0196][0197]
方法一:将式(i)所示化合物(7.05kg,含有乙酸丙酯和正庚烷溶残)溶于丙酮(11.3kg)和无水乙醇(5.6kg)的混合溶剂中,继续搅拌0.5h。将其用钛棒过滤器过滤,然后用丙酮(5.6kg)/无水乙醇(2.8kg)的混合物溶剂淋洗钛棒过滤器,控温20
±
5℃,将滤液缓
慢加入到纯化水(159kg),保温析晶6h。过滤,滤饼用纯化水(18kg)淋洗,然后于60~65℃真空干燥84小时,得到产品6.4kg,产率90.8%,纯度99.74%。
[0198]
方法二:将式(i)所示化合物(26.8kg,含有乙酸丙酯和正庚烷溶残)溶于丙酮(83.0kg)中,继续搅拌0.5h,再经钛棒过滤器过滤。滤液于t≤55℃减压浓缩至无明显馏分。向体系中加入丙酮(83.0kg),减压浓缩至无明显馏分,该操作共重复四次,浓缩完毕,降温至25
±
5℃待用。25
±
5℃下,将浓缩残留物稀释于丙酮(即析晶溶剂,83.0kg)中,并搅拌0.5h。控温25
±
5℃,缓慢滴加到纯化水(268.0kg)中,保温析晶12h。过滤,滤饼用纯化水(26.8kg)淋洗,然后于60
±
5℃,真空干燥12h。得到产品23.96kg,产率89.4%,纯度99.80%。
[0199]
方法三:将方法二中的丙酮替换为无水乙醇和丙酮的混合溶剂溶解含有乙酸丙酯和正庚烷溶残的式(i)的所示化合物,用乙醇蒸馏,用乙醇和丙酮作为析晶溶剂,其它步骤参考方法二的纯化方法,得到产物的产率88.9%,纯度99.63%。
[0200]
三种方法的实验结果见表7
[0201]
表7:三种方法的实验结果
[0202][0203]
注明:当乙酸丙酯和正庚烷的溶残总含量小于0.5%,为合格。
[0204]
实验结果表明,采用方法二,即采用丙酮做溶剂进行析晶,可以使式(i)所示化合物中的溶残含量达到合格的标准,并且不会有新杂质生成。
[0205]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
[0206]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
