1.本发明涉及医药领域。更具体地,涉及一种几乎无细胞毒性的钌配合物、其制备、包含其的复合钌纳米颗粒及制备和应用。
背景技术:
2.常见的铂类抗癌药物,如顺铂、奥沙利铂等,在临床应用较为广泛,但其严重的副作用和耐药性仍是一大问题。光活化化疗(photoactivated chemotherapy,pact)是利用光将惰性的前药转化为有活性的抗癌药物,可以通过选择性照射将药物活性限制在肿瘤组织内,从而减少毒副作用。与传统光动力疗法(photodynamic therapy,pdt)不同的是,pact并不依赖于氧气浓度,因此可用于治疗缺氧实体肿瘤。
3.具有弱配位场的ru(ii)配合物能够在光诱导下发生配体解离,所得到的ru(ii)水配合物可能与生物分子(比如dna)发生共价结合,显示出作为pact试剂的潜力。虽然这些ru(ii)配合物的光物理和光化学性质以及在溶液中形成ru-dna共价结合物已经得到了广泛的研究,但具有高效pact活性的ru(ii)配合物仍然很少。此外,一些ru(ii)配合物,如[ru(7-och
3-dppz)(4-och
3-py)4]
2
(7-och
3-dppz=7-甲氧基-二吡啶并[3,2-a:2’,3
’‑
c]吩嗪,4-och
3-py=4-甲氧基吡啶)能够发生光诱导配体解离,且具有良好的细胞摄取水平,但细胞毒性较低,低的细胞核积累水平可能是其pact疗效低的主要原因。
技术实现要素:
[0004]
本发明的一个目的在于提供一种几乎无细胞毒性的钌配合物,该钌配合物可用于制备细胞核靶向的复合钌纳米颗粒材料,其可用作具有高细胞毒性的高效的光活化化疗试剂并且常氧及乏氧情况下都可以显著抑制肿瘤细胞增殖。
[0005]
本发明的第二个目的在于提供一种几乎无细胞毒性的钌配合物的制备方法。
[0006]
本发明的第三个目的在于提供一种细胞核靶向的复合钌纳米颗粒。该复合钌纳米颗粒具有高效的光活化化疗活性,常氧及乏氧情况下都可以显著抑制肿瘤细胞增殖。钌配合物本身几乎不具备光活化抗肿瘤活性,该复合钌纳米颗粒中,钌配合物被负载到载体c5n2纳米颗粒上后,得到细胞核靶向的复合钌纳米颗粒材料,其可用作具有高细胞毒性的高效的光活化化疗试剂并且常氧及乏氧情况下都可以显著抑制肿瘤细胞增殖。
[0007]
本发明的第四个目的在于提供一种细胞核靶向的复合钌纳米颗粒的制备方法。
[0008]
本发明的第四个目的在于提供一种细胞核靶向的复合钌纳米颗粒的应用。
[0009]
为达到上述第一个目的,本发明采用下述技术方案:
[0010]
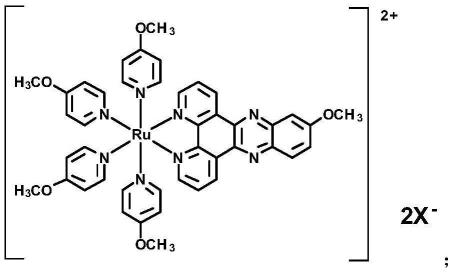
一种几乎无细胞毒性的钌配合物,具有如下式i所述的结构:
[0011][0012]
其中:x-表示平衡电荷的一价阴离子。
[0013]
进一步地,所述x-选自no
3-或pf
6-中的一种。
[0014]
为达到上述第二个目的,本发明采用下述技术方案:
[0015]
一种几乎无细胞毒性的钌配合物的制备方法,包括如下步骤:
[0016]
将摩尔比1:1~1.5的4-甲氧基-邻苯二胺和1,10-邻二氮杂菲-5,6-二酮于溶剂中加热回流反应,反应完后,除去溶剂,重结晶,得7-甲氧基-二吡啶并[3,2-a:2’,3
’‑
c]吩嗪;
[0017]
将摩尔比1:2的二氯苯基钌(ii)二聚体和所述7-甲氧基-二吡啶并[3,2-a:2’,3
’‑
c]吩嗪于溶剂中加热回流反应,除去溶剂,加入六氟磷酸铵饱和溶液,经抽滤、提纯,得所述钌配合物。
[0018]
进一步地,所述提纯的条件为硅胶柱层析,洗脱剂为乙腈和饱和硝酸钾水溶液,其体积比为20~10:1。
[0019]
进一步地,提纯后,向提硅胶柱层析提纯后的化合物中加入一价阴离子的氯化钠溶液或六氟磷酸铵溶液。
[0020]
进一步地,所述溶剂各自独立地选自甲醇或乙醇。
[0021]
为达到上述第三个目的,本发明采用下述技术方案:
[0022]
一种细胞核靶向的复合钌纳米颗粒,包含c5n2纳米颗粒以及负载在所述c5n2纳米颗粒上的如上所述的钌配合物。
[0023]
进一步地,所述c5n2纳米颗粒的粒径为20-25nm;所述复合钌纳米颗粒的粒径为26-30nm。
[0024]
进一步地,所述钌配合物通过静电吸附的作用与c5n2纳米颗粒结合。
[0025]
进一步地,所述c5n2纳米颗粒可根据文献w.chen,j.liu,y.wang,c.jiang,b.yu,z.sun and l.lu,angew.chem.int.ed.,2019,58,6290-6294.中的方法制备得到。
[0026]
进一步地,所述c5n2纳米颗粒的制备包括如下步骤:
[0027]
按摩尔比为1:1称取1,2,4,5-苯四胺四盐酸盐和六酮环己烷八水合物,冰浴处理,氩气环境下缓缓加入80ml n-甲基吡咯烷酮和0.5ml硫酸,160~200℃反应6~10小时,待体系冷却至室温,加水猝灭反应,经过滤、洗涤、干燥得到黑色粉末。
[0028]
称取适量黑色粉末置于玛瑙研钵中,加入适量1mg/ml pvp水溶液,研磨30分钟后,超声(600w,40khz)处理8小时,以10000rpm的速度离心30分钟除去大颗粒,最后将上清液进行透析(6000-8000d)24小时,冻干得到c5n2纳米颗粒。
[0029]
为达到上述第四个目的,本发明采用下述技术方案:
[0030]
一种细胞核靶向的复合纳米颗粒的制备方法,包括如下步骤:
[0031]
将c5n2纳米颗粒的水溶液与钌配合物溶于有机溶剂后得到的溶液混合,搅拌均匀,透析除去有机溶剂和过量的钌配合物,得到复合钌纳米颗粒溶液,冻干备用。
[0032]
进一步地,所述搅拌均匀是指在37℃剧烈搅拌过夜。
[0033]
进一步地,所述有机溶剂选自甲醇、乙醇、乙腈或二甲基亚砜中的一种。
[0034]
进一步地,所述c5n2纳米颗粒的水溶液与所述钌配合物溶于有机溶剂后得到的溶液的体积比为200~50:1。
[0035]
本发明还保护所述的细胞核靶向的复合钌纳米颗粒在制备光活化化疗药物中的应用。
[0036]
本发明的有益效果如下:
[0037]
本发明中提供的钌配合物[ru(7-och
3-dppz)(4-och
3-py)4]
2
本身几乎不具备光活化抗肿瘤活性,c5n2纳米颗粒具有良好的生物相容性以及细胞核靶向能力。将钌配合物负载到c5n2纳米颗粒上,显著提高了钌配合物的细胞摄取及细胞核靶向水平,因此ru-c5n2复合钌纳米颗粒对包括顺铂耐药细胞株在内的一系列癌细胞均表现出高效的pact活性,这些令人鼓舞的结果可以为高效基于ru(ii)配合物的pact制剂的开发提供指导。
附图说明
[0038]
下面结合附图对本发明的具体实施方式作进一步详细的说明。
[0039]
图1示出ru-c5n2纳米颗粒和细胞核染色剂(hoechst 33342)在a549细胞中的共定位成像。
[0040]
图2示出ru-c5n2纳米颗粒在a549细胞中的摄取及定位情况。
具体实施方式
[0041]
为了更清楚地说明本发明,下面结合优选实施例和附图对本发明做进一步的说明。附图中相似的部件以相同的附图标记进行表示。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0042]
实施例1
[0043]
钌配合物[ru(7-och
3-dppz)(4-och
3-py)4](pf6)2的制备方法,包括如下步骤:
[0044]
将210mg(1mmol)的1,10-邻二氮杂菲-5,6-二酮与140mg(1mmol)的4-甲氧基-1,2-苯二胺溶于甲醇中,回流3小时,反应冷却后抽滤,将抽滤物通过乙醇重结晶得到配体7-甲氧基-二吡啶并[3,2-a:2’,3
’‑
c]吩嗪(7-och
3-dppz)的固体。分别称取0.10g(0.2mmol)二氯苯基钌(ii)二聚体和125mg(0.4mmol)7-甲氧基-二吡啶并[3,2-a:2’,3
’‑
c]吩嗪并置于50ml三口瓶中,加入20ml甲醇,氩气保护下回流6小时,然后加入10ml(101.3mg,0.6mmol)agno3水溶液,100℃回流4小时,再加入168.9mg(1.0mmol)4-甲氧基吡啶,100℃继续回流4小时。待反应冷却至室温,加热旋蒸除去溶剂得到粗产物。通过硅胶柱提纯,乙腈和饱和硝酸钾水溶液作为流动相。将得到的化合物溶于水中,加入饱和的nh4pf6水溶液,过滤得到暗红色固体,用少量的水和乙醚多次洗涤,然后真空干燥,得到最终产物。
[0045]
配合物[ru(7-och
3-dppz)(4-och
3-py)4](pf6)2已被报道过(w.sun,y.jian,m.zhou,y.yao,n.tian,c.li,j.chen,x.wang and q.zhou,j.med.chem.,2021,64,7359-7370.)。1h nmr(400mhz,cd3cn)δ9.60(dd,j=13.7,8.1hz,2h),8.84(dd,j=11.9,5.3hz,
2h),8.34(d,j=5.8hz,4h),8.24(d,j=9.3hz,1h),8.06
–
7.96(m,4h),7.74
–
7.60(m,8h),7.49(t,j=6.8hz,4h),7.00(t,j=7.0hz,4h),4.04(s,3h)。hr esi
–
ms:[c
39h32
n8oru]
2
,理论值:365.0872,实测值:365.0874。
[0046]
实施例2
[0047]
c5n2纳米颗粒的制备方法如下:将1.0g(3.52mmol)1,2,4,5-苯四胺四盐酸盐和0.73g(2.35mmol)六酮环己烷八水合物置于三口圆底烧瓶中,氩气保护下并冰浴处理,缓缓加入80ml n-甲基吡咯烷酮和0.5ml硫酸,将体系缓慢升温至室温。之后换为油浴加热至180℃反应8小时,待体系冷却至室温,加水猝灭反应。使用聚四氟乙烯膜过滤得到黑色固体,并进一步用甲醇和水提取三天,之后在120℃下真空干燥24小时得到黑色粉末。称取400mg黑色粉末置于玛瑙研钵中,加入1ml(1mg/ml)pvp水溶液,研磨30分钟后,将混合物转移到装有79ml(1mg/ml)pvp水溶液的锥形瓶中,加入20ml超纯水,超声(600w,40khz)处理8小时,以10000rpm的速度离心30分钟除去大颗粒,最后将上清液进行透析(截留分子量6000-8000d)24小时,冻干备用。
[0048]
c5n2纳米颗粒已被报道过(w.chen,j.liu,y.wang,c.jiang,b.yu,z.sun and l.lu,angew.chem.int.ed.,2019,58,6290-6294.)。
[0049]
负载钌配合物的ru-c5n2纳米颗粒的制备方法如下:
[0050]
为了将[ru(7-och
3-dppz)(4-och
3-py)4]
2
负载到c5n2纳米颗粒上,将5ml(0.8mg/ml)c5n2纳米颗粒水溶液和0.1ml(0.2mg/ml)上述钌配合物乙腈溶液混合,37℃剧烈搅拌过夜,通过透析(截留分子量2000d)除去有机溶剂和过量的钌配合物,得到ru-c5n2纳米颗粒溶液,冻干备用。
[0051]
实施例3
[0052]
通过透射电子显微镜测得c5n2纳米颗粒和负载钌配合物的ru-c5n2纳米颗粒的粒径分别是25nm和28nm左右,动态光散射测得流体动力学直径分别约为36.9nm和41.2nm,与透射电镜测试结果相符。能量色散x射线谱仪显示ru-c5n2纳米颗粒样品中存在钌信号,表明[ru(7-och
3-dppz)(4-och
3-py)4]
2
已成功负载在c5n2纳米颗粒上。另外,c5n2纳米颗粒的zeta电位约为-22.3mv,而ru-c5n2纳米颗粒的zeta电位约为7.5mv,电位的变化情况进一步证实了这个结果。
[0053]
实施例4
[0054]
细胞毒性实验
[0055]
通过mtt法检测[ru(7-och
3-dppz)(4-och
3-py)4](pf6)2,c5n2纳米颗粒和ru-c5n2纳米颗粒对多种肿瘤细胞(人肺癌细胞a549,人宫颈癌细胞hela,人卵巢癌细胞skov-3和人顺铂耐药肺癌细胞a549/ddp)增殖抑制作用。将生长状态良好且处于对数期生长的细胞用无菌pbs缓冲液洗涤3次后,加入适量0.25%的胰酶进行消化,加入适量体积培养基终止消化。将液体转移至离心管中以1200rpm离心5分钟,弃去培养基,加入新鲜的培养基重悬,将细胞以每孔1
×
104个细胞的密度接种在96孔板上,置于37℃培养箱培养24小时。将不同浓度的待测物与细胞共培养4小时后,更换培养基。在光毒性研究中,将细胞置于470nm led灯下光照30分钟,放入培养箱中再培养24小时。暗条件组也采用类似的方法。倒掉培养基,加入200μl浓度为0.5mg/ml的3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐(mtt)溶液孵育4小时,吸去液体,每孔加入200μl dmso和甲醇的混合溶液,通过酶标仪测量每孔在570nm处的
吸光度。[ru(7-och
3-dppz)(4-och
3-py)4](pf6)2,c5n2和ru-c5n2纳米颗粒对不同细胞的ic
50
(半数抑制浓度)结果见表1。
[0056]
结果表明:c5n2纳米颗粒即使在浓度高达400μg/ml时,无论是黑暗中还是光照下(470nm,22.5mw/cm2,30分钟)都表现出了良好的生物相容性。[ru(7-och
3-dppz)(4-och
3-py)4](pf6)2也表现出了较低的细胞毒性,ic
50
值分别在200μm(黑暗)和125μm(光照)以上。有趣的是,ru-c5n2纳米颗粒表现出非常有效的抗癌活性,光照下ic
50
值约为0.18~0.29μm(负载ru(ii)试剂的浓度),光毒性指数值(ic
50dark
/ic
50light
)约为137~246。更重要的是,ru-c5n2纳米颗粒在缺氧(3%o2)条件下对a549细胞的光毒性并未减弱,这与ru(ii)pact试剂的不依赖于氧气浓度的作用机制一致。此外,ru-c5n2纳米颗粒对顺铂耐药a549细胞(a549/ddp)也表现出类似的活性。
[0057]
表1[ru(7-och
3-dppz)(4-och
3-py)4](pf6)2,c5n2和ru-c5n2纳米颗粒对不同肿瘤细胞的ic
50
值(μm)
[0058][0059]a钌配合物:[ru(7-och
3-dppz)(4-och
3-py)4](pf6)2;
b 470nm led灯(22.5mw/cm2)光照30分钟;c未检测;d浓度根据钌配合物的浓度而定;
e ru-c5n2纳米颗粒的光毒性指数(ic
50暗
/ic
50光
)。
[0060]
实施例5
[0061]
细胞共定位实验
[0062]
将a549细胞接种到25mm共聚焦培养皿中,置于37℃培养箱培养24小时。加入染料cy5.5负载的ru-c5n2纳米颗粒(5μm,根据钌配合物的浓度而定),与细胞共培养4小时后,加入商用细胞核染色剂hoechst 33342染色30分钟,使用pbs缓冲液洗涤3次后立即放在共聚焦显微镜下分析。结果如图1。
[0063]
结果表明:蓝色来自于细胞核染色剂hoechst 33342的发光,代表细胞核的位置,红色来自于染料cy5.5负载的ru-c5n2纳米颗粒的发光。pearson相关系数经计算为0.85,表明ru-c5n2纳米颗粒被细胞摄取后主要定位于细胞核。
[0064]
实施例6
[0065]
电感耦合等离子质谱摄取实验
[0066]
采用电感耦合等离子体质谱(icp-ms)测定细胞内ru含量及亚细胞器定位,研究细胞对测试药物的摄取情况。将1
×
106个a549细胞接种到25cm2细胞培养瓶中并培养24小时,加入等体积[ru(7-och
3-dppz)(4-och
3-py)4](pf6)2(1μm)和ru-c5n2纳米颗粒(1μm,根据钌配合物的浓度而定)。与细胞共培养4小时后,倒掉培养基,用无菌pbs缓冲液洗涤3次后,加入适量0.25%的胰酶进行消化,再加入适量体积pbs缓冲液收集并且重悬。使用商用细胞核
提取试剂盒提取出细胞核和细胞质部分,每个样品用60%的hno3溶液处理3天,除去溶剂后用2%的hno3溶解,钌含量采用icp-ms法测定。结果如图2。
[0067]
结果表明:[ru(7-och
3-dppz)(4-och
3-py)4]
2
被细胞摄取后主要定位于细胞质,而ru-c5n2纳米颗粒具有更高的细胞摄取和细胞核核积累水平,这可能是ru-c5n2纳米颗粒抗肿瘤活性显著增强的主要原因。
[0068]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。