一种r-( )-2-(4-羟基苯氧基)丙酸的制备方法
技术领域
1.本发明涉及一种r-( )-2-(4-羟基苯氧基)丙酸的制备方法,属于化合物制备技术领域。
背景技术:
2.r-( )-2-(4-羟基苯氧基)丙酸是合成氰氟草酯、炔草酯、高效氟吡甲禾灵等苯氧基丙酸类除草剂的重要中间体。
3.目前文献和专利报道的制备方法主要以对苯二酚作为起始原料来制备r-( )-2-(4-羟基苯氧基)丙酸甲酯,存在原料转化率低,副产物多的缺点。
4.cn202011608317.5中,将苯酚与s-(-)-2-卤代丙酸反应合成r-( )-2-苯氧基丙酸,经氧化后得到r-( )-2-(4-羟基苯氧基)丙酸。该反应利用强氧化试剂直接在苯环上增加羟基,反应条件较为复杂,副产物及废水较多。
5.cn202010216837.5中,采用对苯二酚和d-乳酸为原料,氮气为载气,对苯二酚和d-乳酸完全气化后进入固定床反应器,在负载型杂多酸催化剂的催化下连续气相合成r-( )-2-(4-羟基苯氧基)丙酸。该工艺需要特殊催化剂,制备条件较为苛刻。反应中将对苯二酚完全气化,能耗较高,不适合工业化生产。
6.cn201711334123.9中,先将对苯二酚分批加入到含有有机溶剂的氢氧化钠溶液中,得到对羟基苯酚钠悬浊液;同时将s-(-)-2-氯丙酸溶于有机溶剂,在冰水浴条件下缓慢加入na2co3固体反应,得到s-(-)-2-氯丙酸钠溶液;最后将两种溶液混合,得到的反应液经减压蒸馏、溶解酸化、萃取、减压蒸馏得到r-( )-2-(4-羟基苯氧基)丙酸。该方法仍无法避免过度烷基化反应,杂质较多,同时采用大量有机溶剂,反应体系为非均相状态,反应效果并不理想。
7.申请人在此方面一直在努力研究,已申请过6项发明专利(cn108129303a,cn110105201a,cn110803987a,cn112062671a,cn114057575a,cn114085145a)。目前申请人经进一步研究获得了不同于上述技术方案的研究成果,并以此来申请本发明专利。
技术实现要素:
8.本发明的主要目的是:克服现有技术存在的问题,提供一种r-( )-2-(4-羟基苯氧基)丙酸的制备方法,反应原料转化率高,副产物少,产品纯度高,后处理简便易行,适合工业化生产。
9.本发明解决其技术问题的技术方案如下:一种r-( )-2-(4-羟基苯氧基)丙酸的制备方法,其特征是,包括以下步骤:第一步、在惰性气体保护下,将对羟基苯甲醛加入水中,加入碱溶液,加热后加入式a化合物,保温反应,反应结束后降温,用酸调节ph后,降温析晶,抽滤,漂洗滤饼,干燥,获得r-( )-2-(4-醛基苯氧基)丙酸;其中,所述式a化合物为s-(-)-2-卤代丙酸酯或其衍生物;
式a中,x选自氟、氯、溴、碘,r选自-oh,-och3,-och2ch3,-nh2,na
,k
;第二步、在惰性气体保护下,将r-( )-2-(4-醛基苯氧基)丙酸加入水中,加入碱溶液,加热后滴加氧化剂,滴完后升温并保温反应,反应结束后降温,用酸调节ph后,降温析晶,抽滤,所得固形物为粗品;将粗品用水精制,抽滤,漂洗滤饼,干燥,获得r-( )-2-(4-羟基苯氧基)丙酸成品。
10.该方法中,在惰性气体保护下,将对羟基苯甲醛和s-(-)-2-卤代丙酸酯或其衍生物经williamson醚化水解反应制备获得中间体r-( )-2-(4-醛基苯氧基)丙酸,然后在碱性条件下,将中间体经dakin氧化反应制备获得光学纯r-( )-2-(4-羟基苯氧基)丙酸。与现有技术中采用对苯二酚为起始原料的制备工艺相比,该方法采用对羟基苯甲醛为起始原料,只有一个酚羟基,不会产生过度烷基化杂质,有效避免了杂质的产生;该方法副反应少,后处理简便,产品收率高且产品纯度高,平均收率不低于80%,产品纯度99.0%以上。
11.本发明进一步完善的技术方案如下:优选地,第一步中,所述式a化合物为s-(-)-2-氯丙酸,s-(-)-2-溴丙酸,s-(-)-2-溴丙酸甲酯,s-(-)-2-氯丙酸甲酯,s-(-)-2-氯丙酸乙酯,s-(-)-2-氯丙酰胺,s-(-)-2-溴丙酸乙酯,s-(-)-2-氯丙酸钠,s-(-)-2-氯丙酸钾之一。
12.优选地,第二步中,所述氧化剂为双氧水、过氧乙酸、过氧苯甲酸、过硫酸钠、过硫酸钾、过碳酸钠之一。
13.更优选地,第一步中,所述式a化合物为s-(-)-2-氯丙酸甲酯;第二步中,所述氧化剂为双氧水。
14.采用以上优选方案后,可进一步优化第一步中的式a化合物,以及第二步中的氧化剂。其中,采用双氧水为氧化剂时,反应过程中主要副产物为水和二氧化碳,能进一步有效减少三废的排放,反应工艺绿色环保。
15.优选地,第一步中,对羟基苯甲醛、式a化合物、碱溶液所含碱的当量比为1:1.0~1.5:1.0~2.0;加入碱溶液后,先加热至30℃~70℃再加入式a化合物;保温反应时,反应温度为30℃~70℃,反应时间为5~10小时;当hplc检测对羟基苯甲醛≤1.0%时反应结束;反应结束后降温至环境温度,用酸调节ph≤2后,降温至0℃~5℃析晶。
16.更优选地,第一步中,对羟基苯甲醛、式a化合物、碱溶液所含碱的当量比为1:1.1~1.2:1.4~1.6;加入碱溶液后,先加热至50℃~60℃再加入式a化合物;保温反应时,反应温度为50℃~60℃,反应时间为6~8小时;当hplc检测对羟基苯甲醛≤0.5%时反应结束;以冰水漂洗滤饼;干燥时采用真空干燥。
17.采用以上优选方案,可进一步优化第一步中的主要具体技术特征。
18.优选地,第二步中,r-( )-2-(4-醛基苯氧基)丙酸、氧化剂、碱溶液所含碱的当量比为1:1.5~2.0:2.0~3.0;加入碱溶液后,先加热至30℃~50℃再滴加氧化剂;保温反应时,反应温度为60℃~80℃,反应时间为5~10小时;当hplc检测r-( )-2-(4-醛基苯氧基)丙酸≤1.0%时反应结束;反应结束后降温至环境温度,用酸调节ph≤1后,降温至0℃~5℃析晶。
19.更优选地,第二步中,r-( )-2-(4-醛基苯氧基)丙酸、氧化剂、碱溶液所含碱的当量比为1:1.6~1.8:2.5~2.6;加入碱溶液后,先加热至40℃~45℃再滴加氧化剂;保温反应时,反应温度为70℃~75℃,反应时间为6~8小时;当hplc检测r-( )-2-(4-醛基苯氧基)丙酸≤0.5%时反应结束;粗品用水精制的次数为一次;以冰水漂洗滤饼;干燥时采用真空干燥。
20.采用以上优选方案,可进一步优化第二步中的主要具体技术特征。
21.优选地,在第一步和第二步中,所述惰性气体分别为氮气或氩气;所述碱溶液分别为氢氧化钠溶液、氢氧化钾溶液或氢氧化钙溶液之一或其中至少两种的混合溶液;采用的酸分别为盐酸、硫酸、磷酸、醋酸之一或其中至少两种的组合;第一步中,对羟基苯甲醛加入水时,对羟基苯甲醛与水的重量比为1:2
±
0.5;第二步中,r-( )-2-(4-醛基苯氧基)丙酸加入水时,r-( )-2-(4-醛基苯氧基)丙酸与水的重量比为1:1.5
±
0.5;粗品用水精制时,r-( )-2-(4-醛基苯氧基)丙酸与水的重量比为1:1
±
0.5。
22.更优选地,在第一步和第二步中,所述惰性气体分别为氮气;所述碱溶液分别为液碱;采用的酸分别为盐酸。
23.采用以上优选方案,可进一步优化第一步、第二步的其余具体技术特征。
24.与现有技术相比,本发明将对羟基苯甲醛经醚化水解反应制备中间体r-( )-2-(4-醛基苯氧基)丙酸,继而经dakin氧化反应制备获得光学纯r-( )-2-(4-羟基苯氧基)丙酸成品。本发明制备过程所用原料易得,反应副产物少、产品收率高且产品纯度高,整个工艺的平均收率不低于80%,产品纯度99.0%以上,后处理简便易行,适合工业化生产。
附图说明
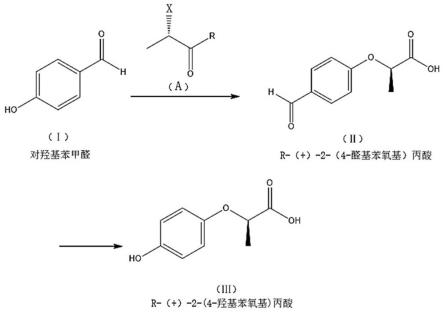
25.图1为本发明的反应原理图。
具体实施方式
26.如图1所示,本发明具体实施的r-( )-2-(4-羟基苯氧基)丙酸的制备方法,包括以下步骤:第一步、在惰性气体保护下,将对羟基苯甲醛加入水中,加入碱溶液,加热后加入式a化合物,保温反应,反应结束后降温,用酸调节ph后,降温析晶,抽滤,漂洗滤饼,干燥,获得r-( )-2-(4-醛基苯氧基)丙酸;其中,式a化合物为s-(-)-2-卤代丙酸酯或其衍生物;
式a中,x选自氟、氯、溴、碘,r选自-oh,-och3,-och2ch3,-nh2,na
,k
;第二步、在惰性气体保护下,将r-( )-2-(4-醛基苯氧基)丙酸加入水中,加入碱溶液,加热后滴加氧化剂,滴完后升温并保温反应,反应结束后降温,用酸调节ph后,降温析晶,抽滤,所得固形物为粗品;将粗品用水精制,抽滤,漂洗滤饼,干燥,获得r-( )-2-(4-羟基苯氧基)丙酸成品。
27.具体而言,第一步中,式a化合物为s-(-)-2-氯丙酸,s-(-)-2-溴丙酸,s-(-)-2-溴丙酸甲酯,s-(-)-2-氯丙酸甲酯,s-(-)-2-氯丙酸乙酯,s-(-)-2-氯丙酰胺,s-(-)-2-溴丙酸乙酯,s-(-)-2-氯丙酸钠,s-(-)-2-氯丙酸钾之一;优选s-(-)-2-氯丙酸甲酯。
28.第二步中,氧化剂为双氧水、过氧乙酸、过氧苯甲酸、过硫酸钠、过硫酸钾、过碳酸钠之一;优选双氧水。
29.具体而言,第一步中:对羟基苯甲醛、式a化合物、碱溶液所含碱的当量比为1:1.0~1.5:1.0~2.0(优选1:1.1~1.2:1.4~1.6);加入碱溶液后,先加热至30℃~70℃(优选50℃~60℃)再加入式a化合物;保温反应时,反应温度为30℃~70℃(优选50℃~60℃),反应时间为5~10小时(优选6~8小时);当hplc检测对羟基苯甲醛≤1.0%时(优选≤0.5%时)反应结束;反应结束后降温至环境温度,用酸调节ph≤2后,降温至0℃~5℃析晶。以冰水漂洗滤饼;干燥时采用真空干燥。
30.具体而言,第二步中:r-( )-2-(4-醛基苯氧基)丙酸、氧化剂、碱溶液所含碱的当量比为1:1.5~2.0:2.0~3.0(优选1:1.6~1.8:2.5~2.6);加入碱溶液后,先加热至30℃~50℃(优选40℃~45℃)再滴加氧化剂;保温反应时,反应温度为60℃~80℃(优选70℃~75℃),反应时间为5~10小时(优选6~8小时);当hplc检测r-( )-2-(4-醛基苯氧基)丙酸≤1.0%时(优选≤0.5%时)反应结束;反应结束后降温至环境温度,用酸调节ph≤1后,降温至0℃~5℃析晶。粗品用水精制的次数为一次;以冰水漂洗滤饼;干燥时采用真空干燥。
31.具体而言,第一步中,对羟基苯甲醛加入水时,对羟基苯甲醛与水的重量比为1:2
±
0.5;第二步中,r-( )-2-(4-醛基苯氧基)丙酸加入水时,r-( )-2-(4-醛基苯氧基)丙酸与水的重量比为1:1.5
±
0.5;粗品用水精制时,r-( )-2-(4-醛基苯氧基)丙酸与水的重量比为1:1
±
0.5。
32.此外,在第一步和第二步中,惰性气体分别为氮气或氩气(优选氮气);碱溶液分别为氢氧化钠溶液、氢氧化钾溶液或氢氧化钙溶液之一或其中至少两种的混合溶液(优选液碱);采用的酸分别为盐酸、硫酸、磷酸、醋酸之一或其中至少两种的组合(优选盐酸)。
33.下面结合实施例对本发明作进一步详细描述。但是本发明不限于所给出的例子。
34.实施例1本实施例为制备r-( )-2-(4-羟基苯氧基)丙酸的一个具体实施示例。
35.本实施例基本过程为上述的本发明具体实施技术方案。
36.本实施例的一些具体细节如下:第一步:制备r-( )-2-(4-醛基苯氧基)丙酸氮气保护下,将对羟基苯甲醛:122g(1.0mol)投入水:244g中,搅拌下加入30%液碱:200g(1.5mol),升温至50℃~60℃,滴加s-(-)-2-氯丙酸甲酯:141g(1.15mol),约2h滴完。s-(-)-2-氯丙酸甲酯滴加完毕,在50℃~60℃下保温反应6~8h,取样中控,对羟基苯甲醛≤0.5%(hplc),反应结束。降至室温,用31%盐酸:295g,调节ph≤2。降温至0℃~5℃析晶30min,抽滤,滤饼用少量冰水:50g漂洗,在40℃~45℃下真空干燥得到中间体,纯度为99.5%,含r-( )-2-(4-醛基苯氧基)丙酸180.6g,收率:93%(理论重量为194.19g)。
37.第二步:制备r-( )-2-(4-羟基苯氧基)丙酸氮气保护下,将r-( )-2-(4-醛基苯氧基)丙酸:180.6g(0.93mol)加入水:271g中,搅拌下加入30%碱液:316g(2.37mol),升温至40℃~45℃,缓慢滴加25%双氧水:215g(1.58mol),约3~4h滴完。双氧水滴加完毕,升温至70℃~75℃,保温反应6~8h,取样分析,r-( )-2-(4-醛基苯氧基)丙酸≤0.5%(hplc),反应结束。降至室温,用31%盐酸:395g调节ph≤1,降温至0℃~5℃析晶,抽滤得粗品:166.5g。
38.将粗品:166.5g投入水:166.5g中,加入活性炭:8g,升温至80℃~85℃脱色1h,趁热抽滤,滤液缓慢降温至0℃~5℃析晶30min,抽滤,滤饼用冰水:50g漂洗,所得湿品在50℃~60℃下真空干燥,得到产品,纯度为99.7%,含r-( )-2-(4-羟基苯氧基)丙酸159.8g,收率94.3%(理论重量为169.46g)。
39.实施例2本实施例为制备中间体r-( )-2-(4-醛基苯氧基)丙酸的一个具体实施示例。
40.本实施例基本过程为上述的本发明具体实施技术方案的第一步。
41.本实施例的一些具体细节如下:氮气保护下,将对羟基苯甲醛:122g(1.0mol)投入水:244g中,搅拌下加入30%液碱:200g(1.5mol),升温至50℃~60℃,滴加s-(-)-2-氯丙酸:125g(1.15mol),约2h滴完。s-(-)-2-氯丙酸滴加完毕,在50℃~60℃下保温反应6~8h,反应结束。降至室温,用31%盐酸:300g,调节ph≤2。降温至0℃~5℃析晶30min,抽滤,滤饼用少量冰水:50g漂洗,在40℃~45℃下真空干燥得到中间体,纯度为99.1%,含r-( )-2-(4-醛基苯氧基)丙酸175.4g,收率:90.3%(理论重量为194.19g)。
42.实施例3本实施例为制备中间体r-( )-2-(4-醛基苯氧基)丙酸的一个具体实施示例。
43.本实施例基本过程为上述的本发明具体实施技术方案的第一步。
44.本实施例的一些具体细节如下:氮气保护下,将对羟基苯甲醛:122g(1.0mol)投入水:244g中,搅拌下加入30%液碱:200g(1.5mol),升温至50℃~60℃,滴加s-(-)-2-溴丙酸甲酯:192g(1.15mol),约2h滴完。s-(-)-2-溴丙酸甲酯滴加完毕,在50℃~60℃下保温反应6~8h,反应结束。降至室温,用31%盐酸:300g,调节ph≤2。降温至0℃~5℃析晶30min,抽滤,滤饼用少量冰水:50g漂洗,在
40℃~45℃下真空干燥得到中间体,纯度为99.0%,含r-( )-2-(4-醛基苯氧基)丙酸180.4g,收率:92.9%(理论重量为194.19g)。
45.实施例4本实施例为制备r-( )-2-(4-羟基苯氧基)丙酸的一个具体实施示例。
46.本实施例基本过程为上述的本发明具体实施技术方案的第二步。
47.本实施例的一些具体细节如下:氮气保护下,将r-( )-2-(4-醛基苯氧基)丙酸:97.1g(0.5mol)加入水:148g中,搅拌下加入30%碱液:170g(1.27mol),升温至40℃~45℃,缓慢滴加40%过硫酸钠溶液:506g(0.85mol),约3~4h滴完。40%过硫酸钠溶液滴加完毕,升温至70℃~75℃,保温反应6~8h。降至室温,用31%盐酸:225g调节ph≤1,降温至0℃~5℃析晶,抽滤得粗品:72.6g。
48.将粗品:72.6g投入水:72g中,加入活性炭:4g,升温至80℃~85℃脱色1h,趁热抽滤,滤液缓慢降温至0~5℃析晶30min,抽滤,滤饼用冰水:25g漂洗,所得湿品在50℃~60℃下真空干燥,得到产品,纯度为99.1%,含r-( )-2-(4-羟基苯氧基)丙酸84.1g,收率92.3%(理论重量为91.1g)。
49.实施例5本实施例为制备r-( )-2-(4-羟基苯氧基)丙酸的一个具体实施示例。
50.本实施例基本过程为上述的本发明具体实施技术方案的第二步。
51.本实施例的一些具体细节如下:氮气保护下,将r-( )-2-(4-醛基苯氧基)丙酸:97.1g(0.5mol)加入水:148g中,搅拌下加入30%碱液:170g(1.27mol),升温至40℃~45℃,缓慢滴加过氧乙酸:65g(0.85mol),约3~4h滴完。过氧乙酸滴加完毕,升温至70℃~75℃,保温反应6~8h。降至室温,用31%盐酸:225g调节ph≤1,降温至0℃~5℃析晶,抽滤得粗品:67.8g。
52.将粗品:67.8g投入水:68g中,加入活性炭:4g,升温至80℃~85℃脱色1h,趁热抽滤,滤液缓慢降温至0℃~5℃析晶30min,抽滤,滤饼用冰水:25g漂洗,所得湿品在50℃~60℃下真空干燥,得到产品,纯度为99.0%,含r-( )-2-(4-羟基苯氧基)丙酸84.3g,收率92.5%(理论重量为91.1g)。
53.对比例1本对比例为制备r-( )-2-(4-羟基苯氧基)丙酸的一个对比示例。
54.本对比例的具体细节如下:第一步:制备r-( )-2-(4-醛基苯氧基)丙酸氮气保护下,将对羟基苯甲醛:122g(1.0mol)投入到水:244g中,搅拌下加入30%液碱:120g(0.9mol),升温至50℃~60℃,滴加s-(-)-2-氯丙酸甲酯:122.5g(1.0mol),约2h滴完。s-(-)-2-氯丙酸甲酯滴加完毕,在50℃~60℃下保温反应6~8h,反应结束。降至室温,用31%盐酸:225g,调节ph≤2。降温至0℃~5℃析晶30min,抽滤,滤饼用少量冰水:50g漂洗,在40℃~45℃下真空干燥得到中间体,纯度为99.1%,含r-( )-2-(4-醛基苯氧基)丙酸167.1g,收率:86%(理论重量为194.2g)。
55.与前文的本发明具体实施技术方案以及实施例1相比,第一步中,30%液碱的加入量过低,导致r-( )-2-(4-醛基苯氧基)丙酸的收率明显下降。
56.第二步:制备r-( )-2-(4-羟基苯氧基)丙酸
氮气保护下,将r-( )-2-(4-醛基苯氧基)丙酸:167.1g(0.86mol)加入到水:250g中,搅拌下加入30%碱液:229g(1.7mol),升温至40℃~45℃,缓慢滴加25%双氧水:150g(1.1mol),约3~4h滴完。双氧水滴加完毕,升温至70℃~75℃,保温反应6~8h。降至室温,用31%盐酸:300g调节ph≤1,降温至0℃~5℃析晶,抽滤得粗品:150.1g。
57.将粗品:150.1g投入水:150g中,加入活性炭:8g,升温至80℃~85℃脱色1h,趁热抽滤,滤液缓慢降温至0℃~5℃析晶30min,抽滤,滤饼用冰水:50g漂洗,所得湿品在50℃~60℃下真空干燥,得到产品,纯度为99.3%,含r-( )-2-(4-羟基苯氧基)丙酸:142.6g,收率91.0%(理论重量为156.7g)。
58.与前文的本发明具体实施技术方案以及实施例1相比,第二步中,30%碱液、25%双氧水的加入量均过低,导致r-( )-2-(4-羟基苯氧基)丙酸的收率明显下降。
59.对比例2本对比例为制备r-( )-2-(4-羟基苯氧基)丙酸的一个对比示例。
60.本对比例的具体细节如下:第一步:制备r-( )-2-(4-醛基苯氧基)丙酸氮气保护下,将对羟基苯甲醛:122g(1.0mol)投入到水:244g中,搅拌下加入30%液碱:280g(2.1mol),升温至50℃~60℃,滴加s-(-)-2-氯丙酸甲酯:196g(1.6mol),约2h滴完。s-(-)-2-氯丙酸甲酯滴加完毕,在50℃~60℃下保温反应6~8h,反应结束。降至室温,用31%盐酸:410g,调节ph≤2。降温至0℃~5℃析晶30min,抽滤,滤饼用少量冰水:50g漂洗,在40℃~45℃下真空干燥得到得到中间体,纯度为98.4%,含r-( )-2-(4-醛基苯氧基)丙酸:178.5g,收率:91.9%(理论重量为194.2g)。
61.与前文的本发明具体实施技术方案以及实施例1相比,第一步中,30%液碱、s-(-)-2-氯丙酸甲酯的加入量均过高,导致r-( )-2-(4-醛基苯氧基)丙酸的纯度明显下降,且收率也有所降低。
62.第二步:制备r-( )-2-(4-羟基苯氧基)丙酸氮气保护下,将r-( )-2-(4-醛基苯氧基)丙酸:178.5g(0.92mol)加入到水:268g中,搅拌下加入30%碱液:374g(2.8mol),升温至40℃~45℃,缓慢滴加25%双氧水:250g(1.84mol),约3~4h滴完。双氧水滴加完毕,升温至70℃~75℃,保温反应6~8h。降至室温,用31%盐酸:450g调节ph≤1,降温至0℃~5℃析晶,抽滤得粗品:162.4g。
63.将粗品:162.4g投入水:163g中,加入活性炭:8g,升温至80℃~85℃脱色1h,趁热抽滤,滤液缓慢降温至0℃~5℃析晶30min,抽滤,滤饼用冰水:50g漂洗,所得湿品在50℃~60℃下真空干燥,得到产品,纯度为99.2%,含r-( )-2-(4-羟基苯氧基)丙酸151.2g,收率90.2%(理论重量为167.6g)。
64.与前文的本发明具体实施技术方案以及实施例1相比,第二步中,30%碱液加入量过高,导致r-( )-2-(4-羟基苯氧基)丙酸的收率明显下降。
65.除上述实施例外,本发明还可以有其他实施方式。凡采用等同替换或等效变换形成的技术方案,均落在本发明要求的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。