1.本发明属于药物化学合成技术领域,具体涉及一种泊沙康唑中间体 2-[(1s,2s)-1-乙基-2-苄氧基丙基]-2,4-二氢-4-[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]-3h-1,2,4-三氮唑-3-酮的制备方法。
背景技术:
[0002]
泊沙康唑(posaconazole,商品名:noxafil),是一类前景广阔的广谱三唑类抗真菌药物,在体外和体内均表现出良好的抗真菌活性,尤其针对一些耐药的菌株。该药物可用于各种复杂罕见真菌感染性疾病的治疗,活性强、起效快,临床上也可作为顽固性侵袭性真菌感染的补救治疗。具有比较理想的安全性和耐受性,肝肾毒性小,适用于长期治疗的患者。
[0003]
如下式所示的泊沙康唑中间体(tm),化学名称为:2-[(1s,2s)-1-乙基-2
‑ꢀ
苄氧基丙基]-2,4-二氢-4-[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]-3h-1,2,4-三氮唑
ꢀ‑
3-酮,主要是由2-[(1s,2s)-1-乙基-2-苄氧基丙基]肼甲醛与[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]氨基甲酸苯酯反应得到。
[0004][0005]
目前,泊沙康唑的中间体(tm)的合成报道相对比较多,主要的问题在于合成路线较长,收率低。其中,在合成2-[(1s,2s)-1-乙基-2-苄氧基丙基]肼甲醛片段中涉及到两个手性中心的合成,光学纯度低也是导致收率低的重要原因之一。此外。传统的合成路线中需要使用到一些反应剧烈的试剂或溶剂,对于工艺条件和设备的要求比较严格,所以导致难以进行工业化生产。
[0006]
专利文献us5625064公开了一种2-[(1s,2s)-1-乙基-2-苄氧基丙基]-2,4-二氢-4-[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]-3h-1,2,4-三氮唑-3-酮中间体制备方法,该方法以l-乳酸甲酯为原料,经胺解、羟基保护、格氏反应、羰基还原、取代、拆分等步骤实现中间体2-[(1s,2s)-1-乙基-2-苄氧基丙基]-2,4-二氢
ꢀ‑
4-[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]-3h-1,2,4-三氮唑-3-酮的合成;但该工艺过程中涉及的步骤较长,其中需要经过拆分步骤,使得收率大大缩减,且在该法过程中使用的对氯苯磺酰氯,反应较为剧烈,对于需要无水、无氧反应条件,条件较为严格,因此不适合工业化实施。
[0007][0008]
专利申请wo2013042138公开了一种由(s)-2-苄氧基丙酸合成n
′‑
[(2s,3s)
ꢀ‑
2-(苄氧基)戊-3-基]甲酰肼和2-[(1s,2s)-1-乙基-2-苄氧基丙基]-2,4-二氢
ꢀ‑
4-[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]-3h-1,2,4-三氮唑-3-酮的方法,工艺方法如下所示。该方法所用到的二氯亚砜试剂有较强的刺激性,更为突出的是其制备过程中要用到价格昂贵并且后处理时很容易着火的二异丁基氢化铝,二异丁基氢化铝反应条件需要在无水条件和超低温下使用,生产危险性大,成本高,操作难度大,难以进行工业化生产。此外,n
′‑
[(2s,3s)-2-(苄氧基) 戊-3-基]甲酰肼的结构含有两个手性中心,专利所用的试剂难以获得的高光学纯度的产品,后处理过程中,还需进行纯化,增加生产成本。
[0009][0010]
为了改善现有技术的缺陷,本发明提供一种泊沙康唑中间体2-[(1s,2s)-1
‑ꢀ
乙基-2-苄氧基丙基]-2,4-二氢-4-[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]-3h-1,2,4
‑ꢀ
三氮唑-3-酮的手性制备方法,其中,对于2-[(1s,2s)-1-乙基-2-苄氧基丙基]肼甲醛中间体的合成可提供两种手性合成方法。所述方法极大的提高了产物反应收率和光学纯度,缩短了工艺合成路线,并且还可将催化剂回收重复使用,不仅减少三废、节约成本,是一种绿色化学合成方法。另外,本发明提供的合成工艺路线短,副产物少,由此获得的产物选择性高,收率较现有技术有较大提升。
技术实现要素:
[0011]
本发明的目的是提供一种泊沙康唑中间体2-[(1s,2s)-1-乙基-2-苄氧基丙基]-2,4-二氢-4-[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]-3h-1,2,4-三氮唑-3-酮的手性制备方法。本发明技术人员创造性的设计合成新的中间体,并利用该中间体可以通过两种方法制备得到2-[(1s,2s)-1-乙基-2-苄氧基丙基]肼甲醛,再与 [4-[4-(4-羟基苯基)-1-哌嗪基]苯基]氨基甲酸苯酯通过对接和关环两步反应得到目标化合物。本发明提供的制备泊沙康唑中间体的方法可以极大的提高产物反应收率和光学纯度,缩短了工艺合成路线,还可将
溶剂和催化剂回收重复使用,减少三废、节约成本,是一种绿色化学合成方法。此外,本发明合成工艺已经过工业生产验证,成本和产品质量较现有技术有较大的优势。
[0012]
本发明的目的通过如下技术方案实现:
[0013]
第一方面,本发明提供一种泊沙康唑中间体的制备方法,所述泊沙康唑中间体为2-[(1s,2s)-1-乙基-2-苄氧基丙基]-2,4-二氢-4-[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]-3h-1,2,4-三氮唑-3-酮(tm),通过采用2-[(1s,2s)-1-乙基-2-苄氧基丙基]肼甲醛与化合物a在碱性条件下缩合得到1-((2s,3s)-2-(苄氧基)戊基-3
‑ꢀ
基)-2-甲酰基-n-(4-(4-(4-羟基苯基)哌嗪-1-基)苯基)肼-1-甲酰胺,再在酸性条件下环化得到泊沙康唑中间体(tm),所述化合物a为[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]氨基甲酸苯酯,反应过程如下:
[0014][0015]
其特征在于,所述2-[(1s,2s)-1-乙基-2-苄氧基丙基]肼甲醛是以式ⅰ所示化合物为起始原料制备得到;
[0016][0017]
其中,r1、r2独立的选自甲基、乙基、丙基、异丙基、叔丁基、环己基、苯基、苄基。
[0018]
在本发明的具体实施方式中,所述r1、r2为甲基。
[0019]
优选的,所述的缩合反应中碱性条件使用的碱包括氢氧化钠、氢氧化钾、磷酸钾、磷酸氢钾、碳酸钾、碳酸钠、三乙胺、n,n-二异丙基乙胺、n-甲基吗啉、咪唑中的一种或两种以上的组合。
[0020]
优选的,所述的环化反应中酸性条件使用的酸包括盐酸、硫酸、磷酸、甲烷磺酸、三氟甲磺酸、氢溴酸、对甲苯磺酸中的一种或两种以上的组合。
[0021]
优选的,所述的缩合反应和环化反应所用溶剂选自二氯甲烷、甲苯、氯苯、四氢呋喃、甲基四氢呋喃、1,4二氧六环、乙二醇二甲醚、乙二醇二乙醚、甲醇、乙醇中的一种或两种以上的组合。
[0022]
优选的,所述2-[(1s,2s)-1-乙基-2-苄氧基丙基]肼甲醛通过如下任一方法制备得到:
[0023][0024]
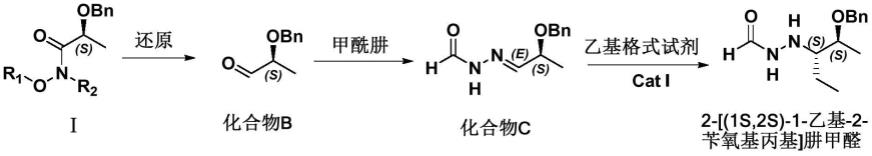
a)式ⅰ化合物经还原得到的醛类化合物b,化合物b与甲酰肼反应得到腙类化合物c,化合物c在催化剂cat i作用下与乙基格式试剂反应生成 2-[(1s,2s)-1-乙基-2-苄氧基丙基]肼甲醛;
[0025][0026]
b)式ⅰ化合物与乙基格式试剂选择性加成后得到的酮类化合物d,化合物d与甲酰肼反应得到腙类化合物e,化合物e在催化剂cat ii作用下与负氢还原剂反应生成2-[(1s,2s)-1-乙基-2-苄氧基丙基]肼甲醛。
[0027]
在制备方法a中,优选的,所述催化剂cat i选自zn(otf)2、sc(otf)3、 cu(otf)2、sn(otf)2、al(otf)2、y(otf)3、ag(otf)2中的一种或两种以上的组合。本发明所述的otf即三氟甲磺酸。
[0028]
进一步的,化合物c与催化剂cat i的摩尔比为1:0.05-0.2。
[0029]
在制备方法b中,优选的,所述催化剂cat ii是配体与金属盐形成的螯合物,所述金属盐可选自氯化亚铜、氯化亚铁、氯化锌、氯化钌、氯化镍、氯化铑、醋酸铜、醋酸铁、醋酸钯中的一种或两种以上的组合。
[0030]
所述配体为式ii所示的化合物:
[0031][0032]
其中,r3、r4独立的选自h、-ch3、-c2h5、叔丁基,c3-8环烷基、苯基、萘基、苄基、对甲氧基苯基、对甲基苯基、对硝基苯基、联苯基、间三甲苯基、苯甲酰基、2,6-二丙基苯基、c3-8杂环烷基、金刚烷基;r5、r6独立的选自h、-ch3、-c2h5、异丙基、叔丁基、苯基、萘基、苄基。
[0033]
优选的,所述配体选自如下化合物中的一种或两种以上的组合。
[0034][0035]
在本发明的最优选实施例中,所述催化剂cat ii是配体-6与氯化亚铜形成的螯合物。
[0036]
所述负氢还原剂选自聚甲基氢硅氧烷(pmhs)。
[0037]
优选的,催化剂cat ii、负氢还原剂与化合物e的摩尔比为1:10-200: 5-100。
[0038]
本发明涉及的乙基格式试剂为ch3ch
2-mgx,x选自cl、br、i。
[0039]
本发明所述的式ⅰ化合物可通过商业途径购买得到,或自行制备得到。在本发明的优选实施例中,所述式ⅰ化合物通过如下方法制备得到:
[0040][0041]
以式ⅲ所示乳酸酯类化合物为起始原料,与式ⅳ所示羟胺盐反应得到化合物f,在溶剂中,化合物f在碱作用下与苄基保护试剂反应得到式ⅰ化合物。
[0042]
其中,r1、r2独立的选自甲基、乙基、丙基、异丙基、叔丁基、环己基、苯基、苄基。
[0043]
r7选自甲基、乙基。
[0044]
y为f、cl、br、i。
[0045]
在本发明的具体实施方式中,式ⅲ化合物为乳酸甲酯,所述r1、r2为甲基
[0046]
优选的,所述溶剂选自二氯甲烷、乙酸乙酯、甲苯、醋酸异丙酯、二氯乙烷、四氢呋喃中的一种或两种以上的组合。
[0047]
所述碱选自碳酸钾、碳酸钠、磷酸钾、磷酸钠、氢氧化钠、氢氧化钾、三乙胺、吡啶中的一种或两种以上的组合。
[0048]
第二方面,本发明提供一种通过如上所述方法制备得到的泊沙康唑中间体(tm)。
[0049]
第三方面,本发明提供一种通过如上所述方法制备得到的泊沙康唑中间体(tm)在制备抗真菌剂泊沙康唑中的用途。
[0050]
第四方面,本发明提供一种式ⅰ所示化合物的制备方法,包括:
[0051][0052]
以式ⅲ所示乳酸酯类化合物为起始原料,与式ⅳ所示羟胺盐反应得到化合物f,在溶剂中,化合物f在碱作用下与苄基保护试剂反应得到式ⅰ化合物。
[0053]
其中,r1、r2独立的选自甲基、乙基、丙基、异丙基、叔丁基、环己基、苯基、苄基。
[0054]
r7选自甲基、乙基。
[0055]
y为f、cl、br、i。
[0056]
在本发明的具体实施方式中,式ⅲ化合物为乳酸甲酯,所述r1、r2为甲基
[0057]
优选的,所述溶剂选自二氯甲烷、乙酸乙酯、甲苯、醋酸异丙酯、二氯乙烷、四氢呋喃中的一种或两种以上的组合。
[0058]
所述碱选自碳酸钾、碳酸钠、磷酸钾、磷酸钠、氢氧化钠、氢氧化钾、三乙胺、吡啶中的一种或两种以上的组合。
[0059]
第五方面,本发明提供一种通过如上所述方法制备得到的式ⅰ所示的化合物。
[0060]
第六方面,本发明提供一种通过如上所述方法制备得到的式ⅰ所示的化合物在制备抗真菌剂泊沙康唑中的用途。
[0061]
本发明提供的泊沙康唑中间体的制备方法具有如下优势:
[0062]
以式ⅰ所示的化合物为重要中间体通过较为温和的条件制备得到 2-[(1s,2s)-1-乙基-2-苄氧基丙基]肼甲醛,再通过缩合和关环两步反应制备得到目标化合物。相较于现有技术,本发明提供的合成方法收率高、路线短,可将溶剂和催化剂回收重复使用,减少三废,节约成本,是一种绿色合成方法。并且,通过优化光学催化剂,使制备的目标化合物光学纯度更高,可达99.99%。
附图说明
[0063]
图1泊沙康唑中间体(tm)的1h核磁共振图谱
[0064]
图2本发明实施例1中化合物2的1h核磁共振图谱
具体实施方式
[0065]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明的部分实施例,而不是全部。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0066]
泊沙康唑中间体的制备
[0067]
实施例1
[0068]
制备路线如下:
[0069][0070]
具体制备方法如下:
[0071][0072]
向
①
号反应釜中加入20kg l-乳酸甲酯,分批加入二甲羟胺盐酸盐(共计 19.7kg),和三乙胺22.56kg加毕后,反应24h,tlc监控,将反应液浓缩干后,加入甲苯40.0kg,加入饱和碳酸氢钠溶液,将ph值调至中性,分液,加入 13.3kg
×
2水洗涤两次,12kg甲苯萃取合并水相,合并有机相干燥后得到化合物1的甲苯溶液约55l。
[0073][0074]
向
②
号反应釜中加入23.34kg甲苯,加入70.7kg碳酸钠,36.4kg氯化苄,将温度降至5℃,缓慢加入上述化合物1的甲苯溶液,控制温度在0~5℃,滴毕,将反应体系升温到15~25℃反应5h后,tlc监控。反应结束,降温至0~5℃,再将25kg水滴加到反应体系中,控制
反应温度小于25℃,分层,甲苯相再用饱和碳酸氢钠(2
×
1.0v/w)洗涤两次后,再用饱和食盐水(0.4 v/w)洗涤1次;分液,合并有机相,干燥后浓缩,得到黄色半固态36.45kg,收率为85.1%,为化合物2,用于下一步反应。
[0075]
化合物2:1h nmr(400mhz,dmso-d6)δ7.37-7.26(m,5h),4.61-4.36(m, 2h),4.01(q,j=6.8hz,1h),3.67(s,3h),2.65(s,3h),1.25(d,j=6.8hz,3h).
[0076][0077]
向
①
反应釜中加入40kg甲苯,3.88kg红铝,氮气置换后氮气保护,降温至-10℃左右,将10kg化合物2用甲苯溶清,缓慢加入反应液中,控制温度在 0~-10℃,滴毕,保温30min,tlc,反应完毕,滴加42.4kg水,随后加入42.4l 盐酸溶液,控制ph值为6,用50kg乙酸乙酯萃取分液,加入15l碳酸氢钠溶液和饱和食盐水15l萃洗后,有机相干燥后浓缩得到化合物3。
[0078][0079]
向
②
号反应釜中加入37.6kg乙醇,5.5kg甲酰肼,15kg化合物3,室温下反应3h,tlc监控,反应结束,浓缩除去乙醇,向浓缩残余物中加入6kg 甲苯,并用10kg
×
2水洗涤两次,分液,有机相干燥后浓缩得到所示化合4。
[0080][0081]
向
③
号反应釜中加入30kg四氢呋喃,中间体10kg的化合物4,1.76kg的 zn(otf)2,氮气保护,室温下搅拌6h,降温至-10~-15℃后,缓慢滴加24.4l 3m 的乙基溴化镁,滴加过程控制反应温度在-5~-10℃,尽量控制在-7~-8℃左右反应。滴毕,保温60~80min。反应结束后,将反应液抽入到盐酸(1.75eq) 中淬灭,控制淬灭温度不超过15℃快速淬灭,萃取分液,水相再用5kg乙酸乙酯反萃取一次,合并有机相加入饱和食盐水(0.5v/m)洗涤一次,干燥后浓缩,得到白色固体9.68kg,收率为84.54%化合物5,手性检测纯度为99.99%。
[0082][0083]
向
①
号反应釜中加入14kg二氯甲烷,5.77kg[4-[4-(4-羟基苯基)-1-哌嗪基] 苯基]氨基甲酸苯酯,2.3kg三乙胺,将3.5kg化合物5用4kg二氯甲烷溶清后滴入反应液中,反应2h。反应结束,加入1m盐酸溶液,将ph值调至中性,分液,有机相干燥后浓缩,得到11.1kg1-((2s,3s)-2-(苄氧基)戊基-3-基)-2-甲酰基-n-(4-(4-(4-羟基苯基)哌嗪-1-基)苯基)肼-1-甲酰胺。
[0084]
向
②
号反应釜中加入13kg 1,4-二氧六环,加入6.5kg上步产物,2.4kg浓盐酸,升温至80℃反应8h,反应完毕,冷却至室温,用饱和碳酸氢钠溶液调节ph值为中性,用乙酸乙酯萃取分液,有机相干燥后浓缩,结晶纯化得到灰白色固体6.09kg,收率为80%,手性纯度在99.99%。
[0085]
经验证,产物的1h nmr(400mhz,cdcl3)δ7.58(s,1h),7.40(d,j=8.9 hz,2h),7.28(s,2h),7.03(d,j=9.0hz,2h),6.94(d,j=8.7hz,2h),6.78(d,j =8.9hz,2h),5.23(s,1h),4.66(d,j=12.0hz,1h),4.43(d,j=12.0hz,1h), 4.21(ddd,j=10.9,6.9,3.9hz,1h),3.90
–
3.75(m,1h),3.50
–
3.34(m,4h), 3.31
–
3.20(m,3h),2.02
–
1.79(m,3h),1.30(d,j=6.3hz,2h),0.91(t,j=7.3 hz,3h).
[0086]
实施例2
[0087]
制备路线如下:
[0088][0089]
以l-乳酸甲酯与二价羟胺盐酸盐制备得到化合物2的过程,以及由化合物5制备得到目标化合物的过程均与实施例1相同。由化合物2制备得到化合物5的制备过程如下:
[0090][0091]
向
④
号反应釜中加入4kg四氢呋喃,3.83kg化合物2,氮气保护,降温至 15~-20℃后,缓慢滴加8.5l3m的乙基溴化镁,滴加过程控制反应温度在-5~-10℃,尽量控制在-7~-8℃左右反应。滴毕,保温60~80min。反应结束后,将反应液抽入到盐酸(1.75eq)中淬灭,控制淬灭温度不超过15℃快速淬灭,萃取分液,水相再用1.5kg乙酸乙酯反萃取一次,合并有机相加入饱和食盐水(0.5v/m)洗涤一次,干燥后浓缩,得到所示化合物6。
[0092][0093]
向
⑤
号反应釜中加入10kg乙醇,0.94kg甲酰肼,3.0kg化合物6,室温下反应3h,tlc监控,反应结束,浓缩除去乙醇,向浓缩残余物中加入1.5kg 甲苯,并用1.5kg
×
2水洗涤两次,分液,有机相干燥后浓缩得到黄色固体为所示化合物7。
[0094][0095]
向
⑥
号反应釜中加入0.37kg氯化亚酮、1.14kg配体-6、1.42kg叔丁醇钠、 14.64kg甲苯氮气置换后1搅拌30min,加入945g pmhs(聚甲基氢硅氧烷),保持温度在20~25℃,将3.66kg化合物7用2.0l甲苯溶清后缓慢滴入,滴毕,反应30min,向反应液中缓慢滴加水,加入醋酸溶液将ph值调至中性,用10l 乙酸乙酯萃取,分液,用1.5l乙酸乙酯萃取水相,合并有机相,干燥后浓缩,得到白色固体3.24kg化合物5,收率为88%,手性纯度为99.99%。
[0096]
实施例2中催化剂catⅱ的筛选优化实验
[0097][0098]
实验目的:通过优化实验筛选得到最适宜本发明的手性催化剂。
[0099]
实验方法:以本发明制备得到的化合物7为起始原料,通过改变催化剂类型制备得到化合物5。
[0100]
实验1
[0101]
向反应瓶中加入10.1g氯化亚铜、3.11g配体-6、41g叔丁醇钠、300ml甲苯氮气置换后搅拌30min,加入2.58g pmhs,保持温度在20~25℃,将100g 化合物7用2.0l甲苯溶清后缓慢滴入,滴毕,反应30min,向反应液中缓慢滴加水,加入醋酸溶液将ph值调至中性,用600ml乙酸乙酯萃取,分液,用 150ml乙酸乙酯萃取水相,合并有机相,干燥后浓缩,得到白色固体88.7g化合物5,收率为87.0%,手性纯度为99.98%。
[0102]
实验2
[0103]
制备原料与制备方法同实验1,区别仅在于将5.53g氯化镍、3.11g配体-6 替换为4.23g氯化亚酮、4.1g配体-1,制备得到白色固体87.2g,收率为87.1%,手性纯度为99.91%。
[0104]
实验3
[0105]
制备原料与制备方法同实验1,区别仅在于将5.53g氯化镍、3.11g配体-6 替换为4.23g氯化亚酮、10.4g配体-4,制备得到白色固体88.24g,收率为87.5%,手性纯度为99.95%。
[0106]
实验4
[0107]
制备原料与制备方法同实验1,区别仅在于将5.53g氯化镍、3.11g配体-6 替换为5.42g氯化亚铁、4.1g配体-1,制备得到白色固体86.8g,收率为86.1%,手性纯度为99.81%。
[0108]
实验5
[0109]
制备原料与制备方法同实验1,区别仅在于将5.53g氯化镍、3.11g配体-6 替换为5.53g氯化镍、3.11g配体-6,制备得到白色固体87.7g,收率为87.0%,手性纯度为99.81%。
[0110]
通过筛选实验发现,只有氯化亚铜与配体-6螯合产物对化合物7的催化作用最佳,产物收率达到88%,产物光学纯度达到99.99%。从机理上分析主要取决于金属离子与卡宾的配位形式以及二者的空间大小对反应底物的影响。
[0111]
对比实施例1
[0112]
参考专利wo 2013/042138公开的方法,以化合物4为起始原料制备化合物5,过程如下:
[0113]
向反应瓶中加入500ml四氢呋喃、200.97g n,o-二甲基甲硅烷基乙酰胺, 100g的化合物4,用250ml甲基叔丁基醚,在室温下搅拌1h。在氮气保护下,将化合物4与n,o-二甲基甲硅烷基乙酰胺的甲基叔丁基醚混合物,在0℃条件下,缓慢滴加24.4l 3m的乙基溴化镁,滴加过程控制反应温度在-5~-10℃,加毕,将反应温度自然升温至室温,搅拌8h。反应结束,用8%的醋酸在0℃的冷水中淬灭反应,随后升温至室温搅拌30min,分液,有机相用饱和食盐水洗涤,分液后,用无水硫酸钠干燥后浓缩得到90.22g浅黄色固有混合物,收率为78.74%,手性纯度为90.33%。
[0114]
通过对比可以发现,在实施例1中,以化合物4为起始原料制备化合物5 的收率为84.54%,产物的手性纯度为99.99%,与对比实施例1相比具有显著优势。因此,按照专利wo 2013/042138公开的方法,完全依靠自身另一个手性来诱导合成手性酰肼化合物5,它的诱导能力是不够的。
[0115]
对比实施例2
[0116]
参考专利cn 102892750公开的方法,以化合物7为起始原料制备化合物 5,过程如下:
[0117]
将100g所得黄色固体化合物7用150ml的甲醇在室温条件下稀释,加入 102g的钯碳(pd/c;5%pd;50%水),进行三次氢气置换,通入氢气的条件下,在室温条件搅拌1-2h,取样送hplc进行检测,过滤反应液,滤饼用200ml 的甲醇淋洗,合并滤液浓缩后得到浅黄色固油混合物,得到粗品95.7g,收率为95%,手性纯度为60%。
[0118]
如上所述,专利cn102892750公开的方法对于亚胺类还原手性控制主要是通过钯碳还原控制所得,钯碳和氢气的条件不仅价格昂贵和危险性系数高。而且通过钯碳加氢还原机理可知,对于亚胺还原的手性控制钯碳加氢是无法诱导控制手性的,故导致产物的手性纯度较低为95%(本发明提供的方法获得的产物手性纯度为99.99%)。此外,钯碳对化合
物7的含量要求比较高,反应体系如果含有微量水或其他残留溶剂或者前面步骤带来的微量金属离子均会毒化钯碳,使得反应转化率降低进而使原料剩余较多,导致收率降低。
[0119]
对比实施例3
[0120]
参考专利cn 106986787公开的方法,以化合物7为起始原料制备化合物 5,过程如下:
[0121]
将所得100g的化合物7用500ml异丙醇溶清加入到反应釜中,随后加入 3g催化剂rh-[(ⅰ)-(r,r)-bdpch]
24
,冷却至0℃左右,将20g硼氢化钠用异丙醇溶清,缓慢滴加至反应液中,控制反应温度在5℃左右,反应2-3h。反应完毕,向体系滴加丙酮,控温在15摄氏度左右,将体系溶剂除干,向浓缩残留物中加入水,用乙酸乙酯萃取,分液,有机相用饱和食盐水洗涤,分液后干燥浓缩得到77.34g浅黄色油状物以及白色固体混合物,收率为76.68%,手性纯度为98.0%。
[0122]
专利cn106986787对于亚胺类还原手性控制主要是由手性催化剂rh-[(ⅰ)-(r,r)-bdpch]
24
来控制,虽然可以将手性纯度提高到98%,但手性铑复合物催化剂价格较高,生产成本难以控制。
[0123]
对比实施例4
[0124]
参考专利us 2014/0343285公开的方法,以化合物5为起始原料制备得到目标化合物泊沙康唑中间体(tm),过程如下:
[0125]
向反应釜中加入91g的化合物5,1000ml二氧六环,100g的[4-[4-(4-羟基苯基)-1-哌嗪基]苯基]氨基甲酸苯酯,升温至95℃后,缓慢加入52g三乙胺,加毕反应24h,反应结束,将反应液过滤,用乙酸乙酯淋洗滤饼,向滤液中加入水,分层后,用乙酸乙酯萃取水相,合并有机相,用2%的氢氧化钠溶液洗涤,随后用5%的盐酸和5%的碳酸氢钠溶液各洗涤一次,干燥后浓缩得到55g 黄色油状物,向油状物中加入150ml异丙醇,将混合液冷却至25℃,搅拌6h 后,过滤,用异丙醇淋洗滤饼,得到46.5g灰白色固体,收率为35.25%,手性纯度为98.11%。向46.5g灰白色固体中加入1.5l甲醇,升温至60℃,随后降温至室温搅拌30min,过滤,干燥,得到34.8g灰白色固体,收率为75%,手性纯度为98.83%。
[0126]
专利us 2014/0343285通过化合物5与[4-[4-(4-羟基苯基)-1-哌嗪基]苯基] 氨基甲酸苯酯一步反应最终获得目标化合物,虽然反应路线短,但是缺陷是目标化合物的收率较低,仅有35%左右。通过研究反应机理发现,获得目标化合物需要经过缩合和关环两个过程获得,且反应原料[4-[4-(4-羟基苯基)-1
‑ꢀ
哌嗪基]苯基]氨基甲酸苯酯在高温强碱下不稳定,所以应该避免长时间在强碱性环境下反应,而专利us 2014/0343285公开的方法中,[4-[4-(4-羟基苯基)-1
‑ꢀ
哌嗪基]苯基]氨基甲酸苯酯在三乙胺环境下反应24h,这也是导致收率降低的主要原因。
[0127]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。