1.本发明涉及一种中链甘油三酸酯的制备方法。
背景技术:
2.中链甘油三酸酯一般是c6~c12脂肪酸形成的甘油三酯,其在制药领域可在脂肪乳注射液中作为原料药使用。中链甘油三酸酯的制备主要有两类方法,一类是化学合成,通过中链脂肪酸与甘油直接酯化,或通过酯交换的方式制备;另一类是酶催化方法,通过脂肪酶进行催化酯化或酯交换。酶催化法具有反应条件温和的优点,但存在酶残留的问题。化学合成法中,酯交换法存在产品纯度低的缺点,因此作为脂肪乳注射液原料药的注射级中链甘油三酸酯主要采用中链脂肪酸与甘油直接酯化的化学合成的方法。
3.中国专利申请(cn 109836328a)采用磺酸甲酯作为催化剂,甘油过量的比例进行酯化,反应结束多次水洗精制。该方法采用甘油过量,必然会导致中链甘油三酸酯含量偏低,单、双酯偏高。专利(cn 108382722a)采用强酸性阳离子交换树脂作为催化剂,采用物料外循环的方式,提高反应转化率,并通过分子蒸馏提高反应纯度。专利(cn104203896a)采用金属氧化铝或者金属氯化物作为催化剂,并提供了一种采用中链甘油三酸酯作为赋形剂的制剂。
4.上述专利均采用催化剂体系,存在催化剂残留的问题。
5.专利(cn107129430a)采用无催化剂体系,反应过程中控制真空度与温度进行酯化反应,反应结束后通过蒸馏出酸即得产品。该方法采用无催化无溶剂体系,安全,符合绿色化学的理念。但该专利升温速度过快,会导致副产物生成,且精制过于简单,可能会存在脂肪酸残留的问题。
技术实现要素:
6.本发明为了克服现有技术中制备中链甘油三酸酯反应转化率和收率不高的缺陷,提供一种中链甘油三酸酯的制备方法,提高了反应转化率和收率,减少了反应副产物的生成、提高了产品纯度,大大降低了脂肪酸的残留,并且本方法操作简单,设备要求低,可实现工业化生产。
7.本发明通过下述技术方案解决上述技术问题。
8.本发明提供了一种中链甘油三酸酯的制备方法,其包括如下步骤:
9.s1、中链脂肪酸和甘油混合,在真空下进行反应;
10.所述反应的温度、真空度及时间控制如下:
11.第一阶段140~150℃,保温保压3~4h,真空度为0.07~0.09mpa;
12.第二阶段160~170℃,保温保压3~4h,真空度为0.07~0.09mpa;
13.第三阶段190~200℃,保温保压8~16h,真空度为0.06~0.08mpa;
14.s2、后处理,即得所述中链甘油三酸酯。
15.步骤s1中,所述第一阶段的真空度较佳地为0.08mpa。
16.步骤s1中,所述第二阶段的真空度较佳地为0.08mpa。
17.步骤s1中,所述第三阶段的真空度较佳地为0.07mpa。
18.其中,所述真空度的控制较佳地为憋压反应,即反应釜与储罐联通,无需持续抽真空。
19.步骤s1中,所述第一阶段的升温方式可为连续升温或分段升温。
20.步骤s1中,所述第二阶段的升温方式可为连续升温或分段升温。
21.步骤s1中,所述第三阶段的升温方式可为连续升温或分段升温。
22.步骤s1中,所述中链脂肪酸可为辛酸、癸酸、c6脂肪酸和c12脂肪酸中的一种或多种。所述c6脂肪酸较佳地为己酸;所述c12脂肪酸较佳地为月桂酸。所述中链脂肪酸较佳地为辛酸和癸酸,所述辛酸和所述癸酸的质量比较佳地为0.1:10~10:0.1,更佳地为6:4~8:2。所述中链脂肪酸较佳地为己酸和辛酸,所述己酸和所述辛酸的质量比较佳地为0.1:10~10:0.1。所述中链脂肪酸较佳地为月桂酸和癸酸,所述月桂酸和所述癸酸的质量比较佳地为0.1:10~10:0.1。
23.步骤s1中,所述中链脂肪酸与所述甘油的摩尔比较佳地为大于3:1,较佳地为(3.6~3.9):1。
24.步骤s1中,所述第一阶段的温度较佳地为143~147℃,例如145℃、146℃。
25.步骤s1中,所述第一阶段的保温保压的时间较佳地为3.5h。
26.步骤s1中,所述第二阶段的温度较佳地为163~167℃,例如164℃、165℃。
27.步骤s1中,所述第二阶段的保温保压的时间较佳地为3.5h。
28.步骤s1中,所述第三阶段的温度较佳地为193~197℃,例如195℃、197℃。
29.步骤s1中,所述第三阶段的保温保压的时间较佳地为10~12h。
30.在本发明某些优选实施方案中,步骤s1中,
31.所述反应的温度、真空度及时间控制如下:
32.第一阶段143~147℃,保温保压3h~4h,真空度0.07~0.09mpa;
33.第二阶段163~167℃,保温保压3h~4h,真空度0.07~0.09mpa;
34.第三阶段193~197℃,保温保压8~16h,真空度0.06~0.08mpa。
35.在本发明某一具体实施方案中,步骤s1中,
36.所述反应的温度、真空度及时间控制如下:
37.第一阶段146℃,保温保压4h,真空度0.09mpa;
38.第二阶段164℃,保温保压4h,真空度0.08mpa;
39.第三阶段180℃,保温保压12h,真空度0.07mpa。
40.在本发明某一具体实施方案中,步骤s1中,
41.所述反应的温度、真空度及时间控制如下:
42.第一阶段140℃,保温保压3h,真空度0.07mpa;
43.第二阶段170℃,保温保压3h,真空度0.07mpa;
44.第三阶段200℃,保温保压8h,真空度0.06mpa。
45.在本发明某一具体实施方案中,步骤s1中,
46.所述反应的温度、真空度及时间控制如下:
47.第一阶段150℃,保温保压4h,真空度0.07mpa;
48.第二阶段160℃,保温保压4h,真空度0.07mpa;
49.第三阶段190℃,保温保压16h,真空度0.09mpa。
50.在本发明某一具体实施方案中,步骤s1中,
51.所述反应的温度、真空度及时间控制如下:
52.第一阶段145℃,保温保压3h,真空度0.08mpa;
53.第二阶段170℃,保温保压3h,真空度0.07mpa;
54.第三阶段200℃,保温保压10h,真空度0.06mpa。
55.在本发明某一具体实施方案中,步骤s1中,
56.所述反应的温度、真空度及时间控制如下:
57.第一阶段140℃,保温保压3.5h,真空度0.09mpa;
58.第二阶段165℃,保温保压3.5h,真空度0.08mpa;
59.第三阶段195℃,保温保压12h,真空度0.07mpa。
60.本发明中,所述步骤s2中,所述后处理可采用本领域常规的方法进行,一般包括:
61.s21、蒸馏。
62.步骤s21中,所述蒸馏可为减压蒸馏。
63.步骤s21中,所述蒸馏的温度可为160~180℃,较佳地为165~175℃。
64.步骤s21中,所述蒸馏的真空度可为≤-0.09mpa;其中,所述真空度控制可为持续抽真空。
65.步骤s21中,所述蒸馏的时间可为2~6h,较佳地为3~5h。
66.步骤s21中,所述蒸馏可去除脂肪酸。
67.较佳地,所述后处理还包括:
68.s22、中和,洗涤;
69.s23、浓缩,过滤。
70.步骤s22中,所述中和可加入碱液中和。所述碱液可为氢氧化钾溶液或氢氧化钠溶液。所述碱液的加入量根据所述蒸馏后得到的所述中链甘油三酸酯的酸值计算。所述酸值的定义为用于中和1g中链甘油三酸酯中全部酸性组分所需要的碱(以koh计)的毫克数,用mgkoh/g表示。
71.较佳地,以所述碱液中碱的质量来计,所述碱液的加入量为中和所述中链甘油三酸酯的理论碱量的1.2~2.0倍,较佳地为1.2~1.5倍。所述碱液的加入量具体计算为:若得到所述中链甘油三酸酯的粗品为m kg,检测其酸值为a mgkoh/g,当所用中和碱液为koh时,以koh的质量计,所述碱液的加入量为(1.2~2.0)*m*a g;当中和碱液为naoh时,以naoh的质量计,所述碱液的加入量为(1.2~2.0)*m*a*40/56g。
72.较佳地,所述碱液的体积为所述中链甘油三酸酯体积的0.5~3倍。
73.步骤s22中,所述中和的温度可为50~80℃,较佳地为60~70℃。
74.步骤s22中,所述中和的时间可为15~60min,较佳地为20~50min,例如25min。
75.步骤s22中,所述洗涤可加入纯化水进行洗涤。所述纯化水的单次体积量较佳地为待洗涤中链甘油三酸酯的体积的0.5~3倍,洗涤次数较佳地为2~4次。
76.步骤s22中,所述洗涤的温度可为50~80℃,较佳地为60~70℃,例如60℃。
77.步骤s22中,所述洗涤的时间可为15~60min,较佳地为20~50min,例如25min。
78.步骤s22中,所述中和可去除剩余脂肪酸,洗涤可除去过量中和液。
79.步骤s23中,所述浓缩可为减压浓缩。
80.步骤s23中,所述浓缩的温度可为70~100℃,较佳地为75~90℃,例如80℃。
81.步骤s23中,所述浓缩的真空度可≤-0.07mpa。
82.步骤s23中,所述过滤可采用滤芯过滤器;其中,所述滤芯过滤器的滤芯的孔径可为0.2~50μm。所述滤芯过滤器的滤膜可为有机滤膜,所述有机滤膜的材质可为尼龙、聚偏氟乙烯、聚四氟乙烯或聚醚砜;所述有机滤膜较佳地为微孔有机滤膜。
83.步骤s23中,所述过滤较佳地为二级过滤;其中,第一级过滤的滤芯的孔径可为0.2~50μm;第二级过滤的滤芯的孔径可为0.2~0.45μm,例如0.2μm或0.45μm。
84.步骤s23中,所述浓缩可除水;所述过滤可除菌、过滤微生物,使微生物、内毒素等指标满足注射剂产品需求。
85.本发明还提供了一种中链甘油三酸酯,其由上述制备方法制得。
86.本发明提供了一种脂肪乳注射液,其包括由上述制备方法制得的中链甘油三酸酯。在所述脂肪乳注射液中,所述中链甘油三酸酯作为营养药用于肠外营养补充。
87.本发明提供了一种油溶性溶剂,其包括由上述制备方法制得的中链甘油三酸酯。所述油溶性溶剂用作药用辅料。
88.在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
89.本发明所用试剂和原料均市售可得。
90.本发明的积极进步效果在于:
91.本发明的制备方法根据反应阶段控制不同的反应真空度、温度及时间,避免了反应初期温度高而导致甘油的聚合,也避免了反应过程中脂肪酸的损失,提高了反应转化率和收率,减少了反应副产物的生成、提高了产品纯度,大大降低了脂肪酸的残留,真实转化率高达98%以上;反应副产物为水,无毒无害。该方法工艺操作简单,设备要求低,可实现工业化生产。
92.本方法制备过程中的过量脂肪酸可回收使用,符合绿色化学的理念。进一步地,采用微孔有机滤膜进行除菌过滤,使中链甘油三酸酯中的微生物、内毒素等指标满足注射剂产品需求。
具体实施方式
93.下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
94.实施例1
95.s1、向酯化釜中投入辛酸1800g,癸酸1200g,甘油550g,抽真空至-0.09mpa以下,切断储罐与真空泵间连接,保持反应釜与储罐间连通。
96.连续升温至146℃,氮气调节真空度至0.09mpa,保温保压反应4h。
97.继续连续升温至164℃,保持真空度0.08mpa,保温保压反应4h。
98.继续连续升温至197℃,调节真空度0.07mpa,保温保压反应12h。
99.s2、后处理:
100.s21、打开储罐与真空泵间的连接,开始抽真空减压蒸馏,180℃蒸馏2h;减压蒸馏的真空度为0.09mpa,降温至60℃,得中链甘油三酸酯粗品2960g。
101.s22、取样测粗品的酸值为9mgkoh/g。加入碱液1500g(其中氢氧化钠的量为计算值的1.5倍),60℃保温搅拌25min,静置分液,弃去下层水相。纯化水洗涤油相两次,每次用量1500g,油相的体积为3000ml,60℃保温搅拌25min,分液。
102.s23、80℃减压浓缩除水,真空度-0.08mpa。浓缩结束,降温至40℃以下,经50μm 0.2μm两级聚醚砜有机滤膜过滤,得中链甘油三酸酯2731g,反应转化率99%,真实收率91.3%。经检测,羟值3,酸值0.05mgkoh/g,不皂化物0.06%。此处“50μm 0.2μm”表示第一级过滤的滤芯的孔径为50μm;第二级过滤的滤芯的孔径为0.2μm。
103.最后制得的中链甘油三酸酯的纯度数据为,中链甘油三酸酯含量99.3%,中链甘油二酸酯含量0.5%,中链甘油单酸酯0.06%。
104.对比例1
105.s1、向酯化釜中投入辛酸1800g,癸酸1200g,甘油550g,抽真空至-0.05mpa以下,切断储罐与真空泵间连接,保持反应釜与储罐间联通。
106.连续升温至145℃,氮气调节真空度至0.05mpa,保温保压反应4h。
107.继续连续升温至165℃,保持真空度0.05mpa,保温保压反应4h。
108.继续连续升温至195℃,调节真空度0.04mpa,保温保压反应13h。
109.s2、后处理:
110.s21、打开储罐与真空泵间的连接,开始抽真空减压蒸馏,170℃蒸馏3h;减压蒸馏的真空度为0.09mpa,降温至50℃,得中链甘油三酸酯粗品2976g。
111.s22、取样测粗品的酸值为9mgkoh/g。加入碱液1200g(其中氢氧化钠的量为计算值的1.2倍),50℃保温搅拌30min,静置分液,弃去下层水相。纯化水洗涤油相两次,每次用量6000g,油相的体积为3000ml,55℃保温搅拌25min,分液。
112.s23、85℃减压浓缩除水,真空度-0.08mpa。减压浓缩3h,浓缩结束,降温至40℃以下,经ptfe(聚四氟乙烯)滤膜过滤,得中链甘油三酸酯2632g,反应转化率92%,真实收率88%。经检测,羟值18,酸值0.06mgkoh/g,不皂化物0.2%。
113.对比例2
114.s1、向酯化釜中投入辛酸2100g,癸酸900g,甘油550g,抽真空至-0.09mpa以下,切断储罐与真空泵间连接,保持反应釜与储罐间联通。
115.连续升温至160℃,氮气调节真空度至0.09mpa,保温保压反应4h。
116.继续连续升温至190℃,保持真空度0.08mpa,保温保压反应12h。
117.s2、后处理:
118.s21、打开储罐与真空泵间的连接,开始抽真空减压蒸馏,165℃蒸馏3h;减压蒸馏的真空度为0.09mpa,降温至50℃,得中链甘油三酸酯粗品2890g。
119.s22、取样测粗品的酸值为9mgkoh/g。加入碱液1200g(其中氢氧化钠的量为计算值的1.2倍),50℃保温搅拌45min,静置分液,弃去下层水相。纯化水洗涤油相三次,每次用量4500g,油相的体积为3000ml,50℃保温搅拌30min,分液。
120.s23、90℃减压浓缩除水,真空度-0.09mpa。减压浓缩2h,浓缩结束,降温至40℃以
下,经pvdf(聚偏氟乙烯)滤膜过滤,得中链甘油三酸酯2620g,反应转化率95%,真实收率89.0%。经检测,羟值11,酸值0.08mgkoh/g,不皂化物0.7%。
121.对比例4
122.采用专利(cn107129430a)技术方案实施,采用辛酸2000g,癸酸1000g,甘油500g;真空调至-0.09mpa,缓慢升温至180℃,保温3h;真空随温度升高逐步调整至~-0.06mpa。真空为-0.09mpa,脱除0.5h。具体见表1。
123.对比例5
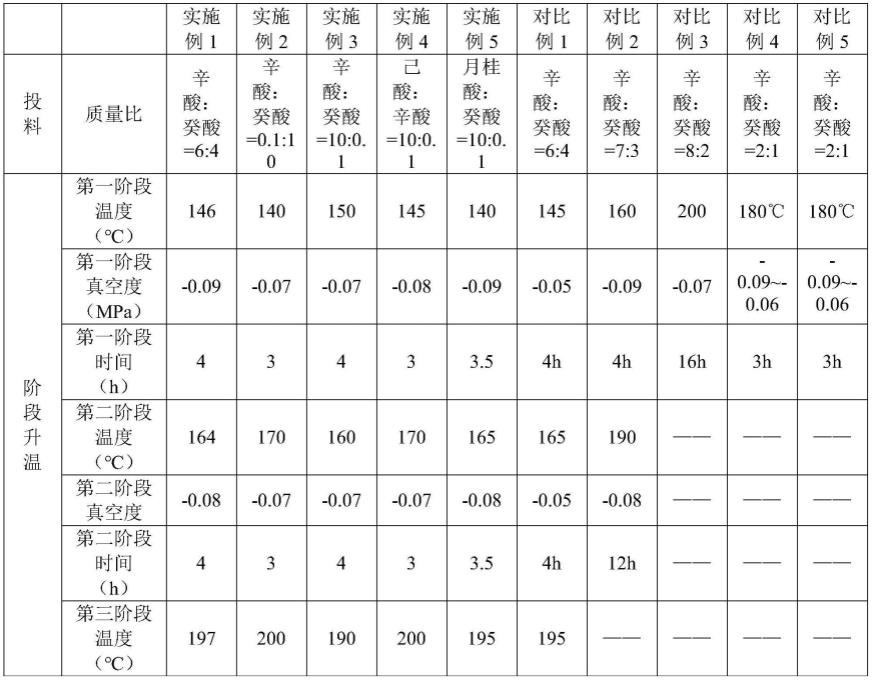
124.采用辛酸2000g,癸酸1000g,甘油500g;升温和蒸馏的操作步骤采用专利(cn107129430a)技术方案,其后处理方式与本技术的后处理工艺实施相同。具体见表1。
125.实施例2~5和对比例3
126.将实施例2~5和对比例3按照实施例1的方法制备,其操作条件和实施例1不同之处见表1,其余条件均和实施例1相同(包括:原料方面,辛酸和癸酸的质量总和为3000g,甘油550g)。
127.表1.实施例1-5和对比例1-3操作条件表
128.[0129][0130]
效果实施例:
[0131]
测得的实施例1-5和对比例1-5的中链甘油三酸酯的纯度数据、真实收率、转化率、羟值、不皂化物和酸值的数据如表2所示。
[0132]
真实收率应当为实际收料量与理论收料量的比值。在本实施例和对比例中脂肪酸是过量的,因此在计算理论收率和转化率时应当以甘油的量计算。理论收料量=(甘油质量/甘油分子量)*产物平均分子量,产物的平均分子量可按照投料的脂肪酸比例近似计算,以实施例1为例,辛酸投料1800g,癸酸投料1200,甘油投料550g。假设反应结束后产物中辛酸癸酸比例保持不变,则产物中辛酸摩尔占比为64.2%,癸酸为35.8%,产品平均分子量为501,理论收料量为2991g,真实收率为91.3%。
[0133]
甘油的消耗质量无法直接获取,可以通过产品的羟值来计算。甘油有3个羟基,通过甘油的质量除以甘油分子量,可计算出反应前羟基的摩尔数;反应后的羟基摩尔数可以通过产品的羟值计算;转化率=(反应前的羟基摩尔数-反应后的羟基摩尔数)/反应前的羟基摩尔数。
[0134]
另外,还测得了中链甘油二酸酯和中链甘油单酸酯的纯度数据。由表2可知,当对比例1采用三阶段升温,但操作条件不在本身的保护范围时,其制得的中链甘油三酸酯的纯度、转化率和真实收率均没有实施例1-5高;当对比例2采用二阶段升温时,其制得的中链甘
油三酸酯的转化率和纯度均没有实施例1-5高;当对比例3采用一步升温时,其制得的中链甘油三酸酯的真实收率、转化率和纯度均没有实施例1-5高。当对比例4采用专利(cn107129430a)的技术实施,其制得的中链甘油三酸酯的真实收率、转化率和纯度均没有实施例1-5高。当对比例5采用专利(cn107129430a)的技术方案,其加上后处理工艺,所采用的后处理方式与本技术的后处理方式实施相同,其制得的中链甘油三酸酯的真实收率、转化率和纯度仍没有实施例1-5高。
[0135]
羟值系指供试品1g中含有的羟基,依据中国药典四部通则中的0713脂肪与脂肪油测定法,进行酰化后,所需氢氧化钾的重量(mg)。羟值表示产品中游离羟基的量,其体现了原料之一甘油的反应程度,羟值越低,甘油所发生的酯化反应越完全。酸值系指用于中和1g中链甘油三酸酯中全部酸性组分所需要的碱(以koh计)的毫克数,用mgkoh/g表示。
[0136]
此处不皂化物代表的是非甘油酯、非脂肪酸类杂质的水平。对比例1-5的羟值均在10以上,实施例1-5的羟值在2-5之间,说明实施例1-5的酯化反应更完全。
[0137]
表2.实施例1-5和对比例1-5的效果数据表
[0138][0139]
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
