1.本发明属于化工领域,具体涉及一种微通道反应器合成高碳羧酸的方法。
背景技术:
2.长链高碳羧酸(碳原子数>8)广泛应用于医药中间体、合成润滑剂中间体、金属加工液、醇酸树脂改性等领域。随着经济的快速发展,其需求量快速增长。长链高碳羧酸制备技术的关键在于氢甲酰化反应与氧化反应。根据氢甲酰化催化剂及其工艺的演变过程,大致可将其分为4代:第一代为1940年德国ruhrchemie公司(现celanese公司)研制的羰基钴或氢羰基钴催化剂;第二代为20世纪60年代shell公司开发的以叔膦配位体改性的钴和铑基催化剂;第三代为20世纪70年代ucc、davymckee(现为kpt)和johnson matthey 3家公司共同开发的三苯基膦(tpp)为配位体的改性铑催化剂和以此为基础开发的“低压羰基合成工艺”(lpo),第四代为80年代ruhrchemie/rhone-poulenc(rch/rp)共同开发的两相工艺及以三苯基膦-三间磺酸盐(tppts)为配位体的水溶性铑催化剂。尽管氢甲酰化催化剂及工艺已发展到第四代,但由于高碳醛/醇的沸点很高,若采用均相工艺,产物与催化剂的分离必须在高温下通过闪蒸,而三代的铑基催化剂在高温下会发生分解而损失。若采用rch/rp的两相催化工艺,因高碳烯烃的水溶性太低,受传质的限制氢甲酰化的反应效率极低。因此,第三代均相铑-膦配合物催化剂和第四代水/有机两相催化剂均未能在高碳烯烃的氢甲酰化中获得应用。迄今为止,85%以上的高碳醇/醛制备仍只能沿用一、二代技术,但这类技术仍存在反应长、反应活性低、催化剂易高温失活和分离困难等一系列问题。此外,目前生产异壬酸的主要工艺是通过异壬醛或醇氧化而制备的,所用到氧气,存在气液反应不充分和燃爆的风险,且有文献报道(organic process research &development,23(8),2019,1709-1717),其氧化反应需48h,产率仅有47%。因此,需要开发新的安全和高效的高碳羧酸的合成工艺。
技术实现要素:
3.为了克服高碳羧酸制备过程中出现的因高碳烯烃原料水溶性低,受传质限制使氢甲酰化反应效率极低,氧化反应效率低并存在燃爆风险等问题,本发明提出了一种微通道反应器合成高碳羧酸的方法。
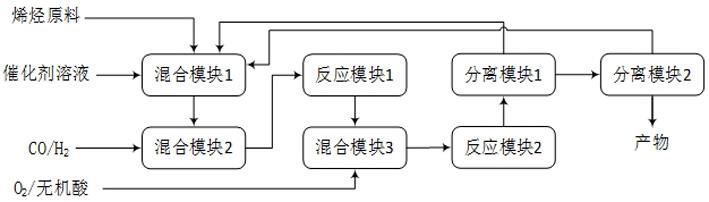
4.为实现上述目的,本发明采用如下技术方案:一种微通道反应器合成高碳羧酸的方法,包括3个混合模块、至少2个反应模块和至少2个分离模块,其是将烯烃原料与催化剂溶液在混合模块1中混合,随后将该物流通入混合模块2中与合成气混合,再将该混合物流通入反应模块1中进行反应,所得反应产物物流与氧气、无机酸溶液在混合模块3中混合,再将所得物流通入反应模块2进行反应,所得反应产物物流随后进入分离模块1进行分液,经分离后的气相排空,所排出的水相物流返回至混合模块1中与原料进行混合,所得到的油相物流进入分离模块2进行精馏,所排出的气相物流返回混合模块1中原料进行混合,所得液相物流即为具有8-17个碳原子的羧酸或二元
羧酸;其中所述混合模块采用微通道混合器,所述反应模块采用微通道反应器,所述分离模块采用微通道分离器。
5.进一步地,所述烯烃原料为碳原子数7-15的高碳烯烃的水溶液,其中烯烃含量为10-100wt.%。
6.进一步地,所述催化剂溶液具体为催化剂的有机溶剂,其浓度为1-500ppm;其中,所述催化剂由金属组分及与其以配位键结合的有机配体按摩尔比0.001-1构成;所述金属组分为co、fe、ni、rh和w中的任意一种或几种,所述有机配体为带有磺酸基的有机膦配体,优选为三苯基膦三间磺酸钠盐(tppts)。
7.进一步地,混合模块1中烯烃原料的进料速度为1-1000ml
·
h-1
;所用催化剂溶液的用量按催化剂与烯烃的摩尔比为0.001-1进行换算。
8.进一步地,混合模块2中合成气的进料速度为0.1-10l
·
h-1
;所述合成气由h2和co组成,两者体积比为0.1-10。
9.进一步地,反应模块1中进行的是氢甲酰化反应,其反应温度为293-623k,反应压力为0.1-25mpa。
10.进一步地,混合模块3中氧气的进料速度为0.1-100l
·
h-1
,氧气纯度>50vol.%,无机酸溶液的浓度>10wt.%,所用无机酸溶液的量按氧气与无机酸的质量比为0.01-1000进行换算;所述无机酸包括硫酸、硝酸、磷酸中的任意一种。
11.进一步地,反应模块2中进行的是氧化反应,其反应条件为293-623k,反应压力为0.1-10mpa。
12.进一步地,各物流在各模块中的停留时间为0.1-30s。
13.进一步地,各模块都连接有进料系统,各物流经进料系统进行传输。
14.本发明的显著优势在于:按本发明方法进行高碳羧酸的合成,不仅提高了反应速率,缩短了反应时间,还降低了催化剂、氧气及无机酸的消耗,且其传质传热效果好,更加安全,通过改变原料烯烃的种类,还可实现各类高碳羧酸的高效连续制备,因此具有良好应用前景。
附图说明
15.图1为本发明合成高碳羧酸的工艺流程图。
具体实施方式
16.如图1所示,本发明微通道反应器合成高碳羧酸的方法是将烯烃原料与催化剂溶液通过进料系统输送到混合模块1中进行混合,随后将其通入混合模块2中与合成气混合,再将该混合物流通入反应模块1进行氢甲酰化反应,所得反应产物物流与氧气和无机酸溶液在混合模块3中混合,再通入反应模块2进行氧化反应,反应产物物流随后进入分离模块1进行分液,经分离后的气相排空,水相物流经进料系统输送至混合模块1中循环使用,所得油相物流进入分离模块2进行精馏,所排出的气相物流经进料系统返回混合模块1中循环使用,所得液相物流即为目标产物。
17.其中所述混合模块采用微通道混合器,所述反应模块采用微通道反应器,所述分
离模块采用微通道分离器。
18.下面结合具体实施例和对比例对本发明的技术方案做进一步的详细说明,但不能以此限定本发明的范围。即凡是依本发明申请专利范围所作的变化与修饰,皆应仍属于发明专利涵盖的范围内。
19.实施例1一种微通道反应器合成高碳羧酸的方法,包括3个混合模块、至少2个反应模块和至少2个分离模块,其是将烯烃原料与催化剂溶液在混合模块1中混合,随后将该物流通入混合模块2中与合成气混合,再将该混合物流通入反应模块1中进行氢甲酰化反应,所得反应产物物流与氧气、无机酸溶液在混合模块3中混合,再将所得物流通入反应模块2进行氧化反应,所得反应产物物流随后进入分离模块1进行分液,经分离后的气相排空,所排出的水相物流返回至混合模块1中循环使用,所得到的油相物流进入分离模块2进行精馏,所排出的气相物流返回混合模块1中循环使用,所得液相物流即为目标产物。
20.所采用的烯烃原料为浓度90wt.%的1-辛烯水溶液;所述催化剂溶液是将羰基钴与有机配体三苯基膦三间磺酸钠盐(tppts)按摩尔比0.1于甲苯中混合制成,其浓度为20ppm。
21.混合模块1中烯烃原料的进料速度为200ml
·
h-1
;所用催化剂溶液的量按与1-辛烯的摩尔比为0.008进行换算。
22.混合模块2中合成气的进料速度为4l
·
h-1
,所述合成气由h2和co组成,h2和co的体积比为3。
23.反应模块1的反应条件为:反应温度为393k,反应压力为13mpa。
24.混合模块3中氧气(纯度>50vol.%)的进料速度为3 l
·
h-1
,所用无机酸溶液为浓度50wt.%的硝酸溶液,其用量按氧气与硝酸的质量比为2进行换算。
25.反应模块2的反应条件为:反应温度为393k,反应压力为2mpa。
26.各物流在各模块中的停留时间为5s,总反应时长为35s。
27.产物中c9羧酸的收率为96.3wt.%,纯度达99%以上。
28.实施例2一种微通道反应器合成高碳羧酸的方法,包括3个混合模块、至少2个反应模块和至少2个分离模块,其是将烯烃原料与催化剂溶液在混合模块1中混合,随后将该物流通入混合模块2中与合成气混合,再将该混合物流通入反应模块1中进行氢甲酰化反应,所得反应产物物流与氧气、无机酸溶液在混合模块3中混合,再将所得物流通入反应模块2进行氧化反应,所得反应产物物流随后进入分离模块1进行分液,经分离后的气相排空,所排出的水相物流返回至混合模块1中循环使用,所得到的油相物流进入分离模块2进行精馏,所排出的气相物流返回混合模块1中循环使用,所得液相物流即为目标产物。
29.所采用的烯烃原料为浓度80wt.%的1-癸烯水溶液;所述催化剂溶液是将羰基镍与有机配体tppts按摩尔比0.05于甲苯中混合制成,其浓度为250ppm。
30.混合模块1中烯烃原料的进料速度为100ml
·
h-1
;所用催化剂溶液的量按与1-癸烯的摩尔比为0.0055进行换算。
31.混合模块2中合成气的进料速度为2l
·
h-1
,所述合成气由h2和co组成,h2和co的体积比为2.5。
32.反应模块1的反应条件为:反应温度为403k,反应压力为15mpa。
33.混合模块3中氧气(纯度>50vol.%)的进料速度为2 l
·
h-1
,所用无机酸溶液为浓度40wt.%的硫酸溶液,其用量按氧气与硫酸的质量比为4进行换算。
34.反应模块2的反应条件为:反应温度为423k,反应压力为1.5mpa。
35.各物流在各模块中的停留时间为10s,总反应时长为70s。
36.产物中c11羧酸的收率为94.6wt.%,纯度达99%以上。
37.实施例3一种微通道反应器合成高碳羧酸的方法,包括3个混合模块、至少2个反应模块和至少2个分离模块,其是将烯烃原料与催化剂溶液在混合模块1中混合,随后将该物流通入混合模块2中与合成气混合,再将该混合物流通入反应模块1中进行氢甲酰化反应,所得反应产物物流与氧气、无机酸溶液在混合模块3中混合,再将所得物流通入反应模块2进行氧化反应,所得反应产物物流随后进入分离模块1进行分液,经分离后的气相排空,所排出的水相物流返回至混合模块1中循环使用,所得到的油相物流进入分离模块2进行精馏,所排出的气相物流返回混合模块1中循环使用,所得液相物流即为目标产物。
38.所采用的烯烃原料为浓度70wt.%的2,2-二甲基壬烯水溶液;所述催化剂溶液是将乙酰丙酮铑与有机配体tppts按摩尔比0.06于甲苯中混合制成,其浓度为50ppm。
39.混合模块1中烯烃原料的进料速度为30ml
·
h-1
;所用催化剂溶液的量按与2,2-二甲基壬烯的摩尔比为0.07进行换算。
40.混合模块2中合成气的进料速度为0.8l
·
h-1
,所述合成气由h2和co组成,h2和co的体积比为1.5。
41.反应模块1的反应条件为:反应温度为383k,反应压力为3.5mpa。
42.混合模块3中氧气(纯度>50vol.%)的进料速度为1.5 l
·
h-1
,所用无机酸溶液为浓度20wt.%的浓硝酸,其用量按氧气与浓硝酸的质量比为1进行换算。
43.反应模块2的反应条件为:反应温度为423k,反应压力为2.5mpa。
44.各物流在各模块中的停留时间为30s,总反应时长为210s。
45.产物中c12羧酸的收率为92.8wt.%,纯度达99%以上。
46.实施例4一种微通道反应器合成高碳羧酸的方法,包括3个混合模块、至少2个反应模块和至少2个分离模块,其是将烯烃原料与催化剂溶液在混合模块1中混合,随后将该物流通入混合模块2中与合成气混合,再将该混合物流通入反应模块1中进行氢甲酰化反应,所得反应产物物流与氧气、无机酸溶液在混合模块3中混合,再将所得物流通入反应模块2进行氧化反应,所得反应产物物流随后进入分离模块1进行分液,经分离后的气相排空,所排出的水相物流返回至混合模块1中循环使用,所得到的油相物流进入分离模块2进行精馏,所排出的气相物流返回混合模块1中循环使用,所得液相物流即为目标产物。
47.所采用的烯烃原料为浓度60wt.%的2,4,4-三甲基-1-戊烯水溶液;所述催化剂溶液是将乙酰丙基铑与有机配体tppts按摩尔比0.05于甲苯中混合制成,其浓度为20ppm。
48.混合模块1中烯烃原料的进料速度为200ml
·
h-1
;所用催化剂溶液的量按与2,4,4-三甲基-1-戊烯的摩尔比为0.002进行换算。
49.混合模块2中合成气的进料速度为2l
·
h-1
,所述合成气由h2和co组成,h2和co的体积比为2.6。
50.反应模块1的反应条件为:反应温度为373k,反应压力为2.4mpa。
51.混合模块3中氧气(纯度>50vol.%)的进料速度为2.4 l
·
h-1
,所用无机酸溶液为浓度15wt.%的硝酸溶液,其用量按氧气与硝酸的质量比为3.6进行换算。
52.反应模块2的反应条件为:反应温度为393k,反应压力为1.5mpa。
53.各物流在各模块中的停留时间为8s,总反应时长为56s。
54.产物中c9羧酸的收率为98.6wt.%,纯度达99%以上。
55.对比例 采用常规高压釜式反应器进行高碳羧酸的合成采用高压釜式反应器,双釜串联,反应釜1进行氢甲酰化反应,反应釜2为氧化反应,后续以分液设备和精馏设备分别作为分离模块1和2,其具体操作为将烯烃原料、催化剂溶液和合成气通入反应釜1,升高至一定压力和温度后进行氢甲酰化反应,所得反应产物物流与氧气、无机酸溶液再通入反应釜2进行氧化反应,所得反应产物物流随后进入分离模块1进行分液,经分离后的气相排空,所排出的水相物流返回至反应釜1中与原料进行混合,所得到的油相物流进入分离模块2进行精馏分离,所排出的气相物流返回混合模块1中原料进行混合,所得液相物流即为目标产物。
56.所采用的烯烃原料为浓度60wt.%的2,4,4-三甲基-1-戊烯水溶液;所述催化剂溶液是将乙酰丙基铑与有机配体tppts按摩尔比0.05于甲苯中混合制成,其浓度为20ppm。
57.反应釜1中烯烃原料的进料量为2l;所用催化剂溶液的量按与2,4,4-三甲基-1-戊烯的摩尔比为0.002进行换算;反应前先通氮气进行置换后,升温至373k,随后采用合成气将反应釜压力提高至2.4mpa,所述合成气由h2和co组成,h2和co的体积比为2.6。
58.反应釜2中通入氧气(纯度>50vol.%)至反应压力为1.5mpa,并加热至反应温度为393k;所用无机酸溶液为浓度15wt.%的硝酸溶液,其用量按氧气与硝酸的质量比为3.6进行换算。
59.反应釜1中反应时长为8h,反应釜2中反应时长为6h。
60.产物中c9羧酸的收率为40wt.%,产品纯度约60%。
61.以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
