1.本发明属于有机合成和药物化学领域,具体涉及一种中药八角枫中的哌啶类生物碱及其提取纯化、半合成方法和应用。
背景技术:
2.人类免疫缺陷病毒(human immunodeficiency virus,hiv)是艾滋病的病原体。自1981年,发现第一例艾滋病患者以来,艾滋病已在世界范围内迅速扩散,成为了一类威胁人类健康的重大传染病。在全世界范围内控制艾滋病的传播,一直所面临着严峻挑战。迄今为止,尚无有效的艾滋病疫苗或能完全治愈艾滋病的药物问世,众多科研人员一直致力于高效低毒的抗hiv药物的寻找和研发。
3.实践证明,中医药目前在治疗艾滋病方面显示出了良好的疗效,能明显改善临床症状,有利于机体抵抗力和免疫调节功能的恢复,且具有费用低、使用方便等优势。由于目前临床上用于抗hiv病毒的药物毒副作用较大,因此从丰富的天然产物中寻找高效低毒的治疗艾滋病药物的研究已成为当前国际医药界的研究热点。
4.随着对hiv认识的不断深入,引进和开发出了许多筛选抗hiv药的分子细胞模型,利用这些模型筛选出了许多有价值的单味中药、复方中药及单一活性成分等,为进一步开发防治艾滋病药物提供了极富价值的物质基础。
[0005][0006]
八角枫alangium chinense(lour.)harms为贵州苗族民间习用治疗炎症的药物植物,通过对其进行分离纯化,得到一个结构新颖的哌啶生物碱类化合物(
±
)-6-(2-(pyridin-3-yl)piperidin-1-yl)-[2,3'-bipyridin]-3-ol,命名为(
±
)-alanginenmine a。对其进行了抗hiv病毒活性测定,结果表明其具有一定的抗hiv活性,ic
50
值为15.1μmol/l。由于(
±
)-alanginenmine a在植物八角枫alangium chinense(lour.)harms含量低,因此在实践中对其深入的开发利用会受到一定程度的限制,通过化学半合成方法制备化合物(
±
)-alanginenmine a,不失为一条方便有效的途径。
技术实现要素:
[0007]
本发明的目的是提供一种中药八角枫中的哌啶类生物碱;
[0008]
本发明的另一目的是提供一种中药八角枫中的哌啶类生物碱提取纯化方法;
[0009]
本发明的另一目的是提供一种中药八角枫中的哌啶类生物碱消旋体的半合成方法;
[0010]
本发明的另一目的是提供一种中药八角枫中的哌啶类生物碱在制备抗hiv药物方
面的应用。
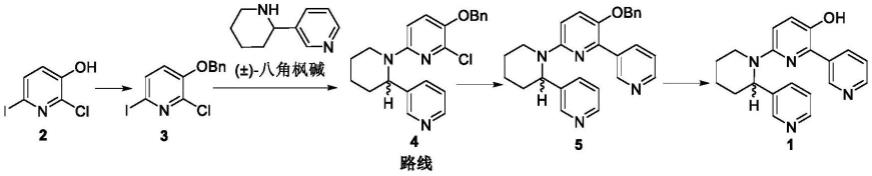
[0011]
本发明所述哌啶类生物碱为
±
)-alanginenmine a。
[0012]
本发明所述(
±
)-alanginenmine a化合物表征为:
[0013]
结构式:
[0014][0015]
分子式:c
20h20
n4o;
[0016]
状态:无定形固体;
[0017]
hresims m/z:333.1707[m h]
(图1)
[0018]
硅胶薄层检识:以比例为110:10:1的二氯甲烷:乙酸乙酯:氨水展开(rf值为0.5),在254nm下有荧光,经碘化铋钾显色显色后呈现红色斑点,经茚三酮显色烘烤呈浅紫色斑点;
[0019]1h-nmr(600mhz,cd3od,图2)、
13
c-nmr(150mhz,cd3od,图3)数据如下:
[0020][0021][0022]
本发明所述吲哚类生物碱的提取纯化方法具体为:
[0023]
取3~12kg干燥的八角枫根,粉碎后用甲醇回流提取2~5次,每次20l,每次1~5小时,得到甲醇提取物,减压除去有机溶剂后,得到醇提浸膏;边搅拌边加入hcl溶液将ph调至1~3,提取物溶解后过滤,再用氨水将滤液的ph值调节至8~12,然后用3~8l的etoac萃取3~5次,得到萃取液,减压浓缩得浸膏,将该浸膏进行硅胶柱层析,用1:0~0:1的二氯甲烷-甲醇溶剂梯度洗脱,得到7个组分a-g;将组分b进行硅胶柱层析,以用1:0~0:1的石油醚-乙酸乙酯溶剂梯度洗脱,得到11个组分b1~b
11
;将b
10
进行sephadex lh-20柱色谱分离,以甲醇做洗脱剂,得到含化合物1的粗品,随后该粗品用mci-gel chp 20p柱分离,以1:4~4:1的甲醇-水作为洗脱剂,梯度洗脱,得到化合物1。
[0024]
优选的,本发明所述吲哚类生物碱的提取纯化方法具体为:
[0025]
取5kg干燥的八角枫根,粉碎后用甲醇回流提取3次,每次20l,每次3小时,得到甲醇提取物,减压除去有机溶剂后,得到醇提浸膏;边搅拌边加入hcl溶液将ph调至2,提取物溶解后过滤,再用氨水将滤液的ph值调节至10,然后用5l的etoac萃取4次,得到萃取液,减压浓缩得浸膏,将该浸膏进行硅胶柱层析,用1:0~0:1的二氯甲烷-甲醇溶剂梯度洗脱,得到7个组分a-g;将组分b进行硅胶柱层析,以用1:0~0:1的石油醚-乙酸乙酯溶剂梯度洗脱,得到11个组分(b1~b
11
);将b
10
进行sephadex lh-20柱色谱分离,以甲醇做洗脱剂,得到含化合物1的粗品,随后该粗品用mci-gel chp 20p柱分离,以1:4~4:1的甲醇-水作为洗脱剂,梯度洗脱,得到化合物1。
[0026]
本发明所述(
±
)-alanginenmine a化合物半合成方法如下:
[0027]
以2-氯-3-羟基-6-碘吡啶和(
±
)-八角枫碱为原料,按以下合成路线合成化合物(
±
)-alanginenmine a;
[0028][0029]
其中,bn为苄基。
[0030]
本发明所述(
±
)-alanginenmine a化合物的半合成方法具体如下:
[0031]
(1)合成化合物3:
[0032]
将化合物2(2-氯-3-羟基-6-碘吡啶)、溴化苄和k2co3混合,加入四氢呋喃,在65℃下搅拌反应4小时后,减压浓缩,硅胶柱层析,得2-氯-3-苄氧基-6-碘吡啶,即化合物3;
[0033]
(2)合成化合物4:
[0034]
氩气保护下,将化合物3、八角枫碱、pd(pph3)4、正丁基二(1-金刚烷基)膦和cs2co3混合,加入1,4-二氧六烷,升温至120℃搅拌反应72小时,加水淬灭,再用乙酸乙酯萃取3~5次,合并有机相,无水硫酸钠干燥,硅藻土过滤,减压浓缩,硅胶柱层析,得3-(benzyloxy)-2-chloro-6-(2-(pyridin-3-yl)piperidin-1-yl)pyridine,即化合物4;
[0035]
(4)合成化合物5:
[0036]
氩气保护下,将化合物4和吡啶-3-硼酸于甲苯和乙醇的混合溶液中溶解,加入cs2co3、k3po4和催化剂pdcl2(pph3)2,在100℃下搅拌反应72小时后,减压浓缩,硅胶柱层析,得3-(benzyloxy)-6-(2-(pyridin-3-yl)piperidin-1-yl)-2,3'-bipyridine,即化合物5;
[0037]
(5)合成化合物1:
[0038]
氩气保护下,取nah(60%in oil)和pd(oac)2分散于二甲基乙酰胺,在室温下搅拌5min,再加入化合物5,在60℃下搅拌反应3小时,加饱和氯化铵水溶液淬灭,用乙酸乙酯萃取3~5次,合并有机相,再用水反萃3~5次,合并有机相,无水硫酸钠干燥,硅藻土过滤,减压浓缩,硅胶柱层析,得6-(2-(pyridin-3-yl)piperidin-1-yl)-[2,3'-bipyridin]-3-ol,即化合物1。
[0039]
本发明半合成方法步骤(1)所述的化合物2:溴化苄:k2co3的摩尔比例为1:1~2:2~4;硅胶柱层析所用洗脱剂为石油醚/乙酸乙酯,石油醚/乙酸乙酯的体积比为30:1~15:1;步骤(2)所述的化合物3、八角枫碱、cs2co3的摩尔比例为1~2:1:2;pd(pph3)4和正丁基二(1-金刚烷基)膦的优选摩尔比例为1:4;硅胶柱层析所用洗脱剂为石油醚/乙酸乙酯,石油醚/乙酸乙酯优选的体积比为3:1。
[0040]
本发明半合成方法步骤(3)需要氩气保护;所述化合物4:吡啶-3-硼酸:cs2co3:k3po4:pdcl2(pph3)2的摩尔比例为1~2:1:1~3:1~3:0.02~0.1;优选反应温度为100℃,硅胶柱层析所用洗脱剂为石油醚/乙酸乙酯,石油醚/乙酸乙酯优选的体积比为1:3。
[0041]
本发明半合成方法步骤(4)需要氩气保护;所述化合物5:nah:pd(oac)2的摩尔比例为1:1~2:0.02~0.1;优选反应温度为60℃,硅胶柱层析所用洗脱剂为二氯甲烷/甲醇/氨水,二氯甲烷/甲醇/氨水优选的体积比为250:26:1。
[0042]
本发明所述的(
±
)-alanginenmine a在抗hiv药物方面的应用,具体为在制备抗hiv药物或药物制剂中方面的应用;所述制剂为加入药学上可接受的辅料按常规工艺制备成药学上可接受的固体制剂或液体制剂;所述固体制剂为颗粒剂、胶囊剂、片剂、丸剂、散剂、冻干粉针剂;所述液体制剂为注射制剂、口服液。
[0043]
本发明所述分离纯化制备的新化合物1的结构鉴定:
[0044]
(
±
)-alanginenmine a(1),hresims检测到在m/z 333.1707[m h]
处有准分子离子峰(图1),将其分子式指定为c
20h20
n4o(理论计算c
20h21
n4o
的结果为:333.1710)。化合物1的1h nmr(图2)、
13
c nmr(图3)和hsqc(图4)波谱数据显示含有10个吡啶-哌啶生物碱骨架的碳峰,包括四个亚甲基(δ
c 48.3、33.1、26.6和22.6)、五个次甲基(δ
c 57.2、125.2、137.3,147.7和149.4)和一个芳香季碳(δ
c 141.9),与八角枫碱的碳谱峰相似,推断化合物1含有八角枫碱的结构片段。其余的10个芳基c(δ
c 155.3、150.4、148.3、146.9、138.7、138.2、136.4、128.6、124.6和113.4)和1个o和2个n,暗示存在羟基取代的双吡啶单元。在该双吡啶结构中,δ
h 8.35/δ
h 7.41/δ
h 8.39的1h-1
h cozy相关性(图6)和δ
h 8.35/δ
c 150.4、δ
h 7.41/δ
c 136.4(季碳c-3”')的hmbc相关性(图5)建立了一个3”'-取代的吡啶(d环)。另一个5
”‑
羟基吡啶基团(c环),通过详细的2d nmr数据分析分配,如化合物2d nmr的相关性图所绘。δ
h 8.35(h-4”')与δ
c 138.7(c-6”)的hmbc相关性进一步确定了c环的6
”‑
位和d环的3”'直接相连,确定了羟基取代的双吡啶结构接连方式。最后通过hmbc光谱中δ
h 5.29(h-2)到δ
c 155.3(c-2”)的相关性,确定了c环和八角枫碱之间的连接位置,为c环的2”位与八角枫碱的n-1位直接相连。因此,化合物1的平面结构被确定为前所未有的吡啶-吡啶-八角枫骨架,如结构式所示。
[0045][0046]
化合物1的关键hmbc1h-1
h cosy和roesy相关。
[0047]
由于c-2位与c-3'之间是柔性单键,无法通过roesy(图7)实验来确定化合物1的相对构型,但δ
h 6.82(h-3”)和δ
h 5.29(h-2)之间的noe相关性,进一步表明5”位接有羟基。我们使用半制备型的hplc(手性柱为oh-h)成功分离得到了两种对映异构体(1a和1b),1a和1b的旋光分别为(c=0.32,甲醇)和(c=0.17,甲醇)。而化合物1的比旋光度值(c=0.10,甲醇),暗示我们所分离的天然产物为消旋体。基于以上所述,化合物1被鉴定为(
±
)-6-(2-(pyridin-3-yl)piperidin-1-yl)-[2,3'-bipyridin]-3-ol,命名为(
±
)-alanginenmine a。
[0048]
本发明具有如下的有益效果:
[0049]
1、本发明通过提取纯化分离得到一个结果新颖的哌啶生物碱类化合物(
±
)-6-(2-(pyridin-3-yl)piperidin-1-yl)-[2,3'-bipyridin]-3-ol,命名为(
±
)-alanginenmine a。
[0050]
2、通过体外抗hiv蛋白酶活性测定结果显示,化合物1显示出对hiv-1蛋白酶的抑制作用,具有一定的抗hiv活性,化合物1在0.1mg/ml、0.01mg/ml和0.001mg/ml(dmso)的给药浓度下,抑制率分别为100%、45.2%和24.1%,化合物1的ic
50
值为15.1μmol/l。
[0051]
3、本发明通过半合成方法,以2-氯-3-羟基-6-碘吡啶和(
±
)-八角枫碱为原料,经四步反应,完成了化合物(
±
)-alanginenmine a的半合成;本发明的半合成方法,中间体稳定,反应容易控制,产率优良,解决了化合物(
±
)-alanginenmine a在植物八角枫含量低导致其深入开发利用受到限制的问题,可以工业化大量制备化合物(
±
)-alanginenmine a。
附图说明
[0052]
图1本发明从八角枫制备的新化合物1的hr-esi-ms图
[0053]
图2本发明从八角枫制备的新化合物1的1h-nmr图(cd3od)
[0054]
图3本发明从八角枫制备的新化合物1的
13
c-nmr图(cd3od)
[0055]
图4本发明从八角枫制备的新化合物1的hsqc图(cd3od)
[0056]
图5本发明从八角枫制备的新化合物1的hmbc图(cd3od)
[0057]
图6本发明从八角枫制备的新化合物1的1h-1
h cosy图(cd3od)
[0058]
图7本发明从八角枫制备的新化合物1的roesy图(cd3od)
[0059]
图8本发明半合成的新化合物1的hr-esi-ms图
[0060]
图9本发明半合成的新化合物1的1h-nmr图(cd3od)
[0061]
图10本发明半合成的新化合物1的
13
c-nmr图(cd3od)
具体实施方式
[0062]
通过下述实施例将有助于进一步理解本发明,但这并不意味着对本发明的任何限制。
[0063]
实施例1
[0064]
哌啶类生物碱的提取纯化方法:
[0065]
取5kg干燥的八角枫根,粉碎后用甲醇回流提取3次,每次20l,每次3小时,得到甲醇提取物;减压除去有机溶剂后,得到醇提浸膏461g,边搅拌边加入hcl溶液将ph调至2,提取物溶解后过滤,再用氨水将滤液的ph值调节至10,然后用5l的etoac萃取4次,得到萃取液,减压浓缩得浸膏40g;将该浸膏进行硅胶柱层析,用1:0~0:1的二氯甲烷-甲醇溶剂梯度洗脱,得到7个组分a-g,其中组分b为20g;组分b进行硅胶柱层析,以用1:0~0:1的石油醚-乙酸乙酯溶剂梯度洗脱,得到11个组分(b1~b
11
),其中b
10
为639mg;b
10
进行sephadex lh-20柱色谱分离,以甲醇做洗脱剂,得到含化合物1的粗品130mg,随后该粗品用mci-gel chp 20p柱分离,以1:4~4:1的甲醇-水作为洗脱剂,梯度洗脱,得到6.69mg的化合物1。
[0066]
化合物表征:
[0067]
化合物1:(
±
)-alanginenmine a
[0068]
结构式:
[0069][0070]
分子式:c
20h20
n4o
[0071]
状态:无定形固体
[0072]
hresims m/z:333.1707[m h]
[0073]
硅胶薄层检识:以比例为110:10:1的二氯甲烷:乙酸乙酯:氨水展开(rf值为0.5),在254nm下有荧光,经碘化铋钾显色剂显色后呈现红色斑点,经茚三酮显色烘烤呈浅紫色斑点;
[0074]1h-nmr(600mhz,cd3od)、
13
c-nmr(150mhz,cd3od)数据如下:
[0075]
[0076][0077]
实施例2
[0078]
取12kg干燥的八角枫根,粉碎后用甲醇回流提取5次,每次5小时,得到甲醇提取物;减压除去有机溶剂后,得到醇提浸膏800g,边搅拌边加入hcl溶液将ph调至3,提取物溶解后过滤,再用氨水将滤液的ph值调节至12,然后用8l的etoac萃取5次,得到萃取液,减压浓缩得浸膏60g;将该浸膏进行硅胶柱层析,用1:0~0:1的二氯甲烷-甲醇溶剂梯度洗脱,得到7个组分a-g,其中组分b为30g;组分b进行硅胶柱层析,以用1:0~0:1的石油醚-乙酸乙酯溶剂梯度洗脱,得到11个组分(b1~b
11
),其中b
10
为900mg;b
10
进行sephadex lh-20柱色谱分离,以甲醇做洗脱剂,得到含化合物1的粗品200mg,随后该粗品用mci-gel chp 20p柱分离,以1:4~4:1的甲醇-水作为洗脱剂,梯度洗脱,得到8mg的化合物1。
[0079]
实施例3
[0080]
取3kg干燥的八角枫根,粉碎后用甲醇回流提取2次,每次1小时,得到甲醇提取物;减压除去有机溶剂后,得到醇提浸膏200g,边搅拌边加入hcl溶液将ph调至1,提取物溶解后过滤,再用氨水将滤液的ph值调节至8,然后用3l的etoac萃取3次,得到萃取液,减压浓缩得浸膏20g;将该浸膏进行硅胶柱层析,用1:0~0:1的二氯甲烷-甲醇溶剂梯度洗脱,得到7个组分a-g,其中组分b为10g;组分b进行硅胶柱层析,以用1:0~0:1的石油醚-乙酸乙酯溶剂梯度洗脱,得到11个组分(b1~b
11
),其中b
10
为400mg;b
10
进行sephadex lh-20柱色谱分离,以甲醇做洗脱剂,得到含化合物1的粗品100mg,随后该粗品用mci-gel chp 20p柱分离,以1:4~4:1的甲醇-水作为洗脱剂,梯度洗脱,得到4mg的化合物1。
[0081]
实施例4
[0082]
半合成制备(
±
)-alanginenmine a:
[0083]
步骤1合成化合物3
[0084]
在配备有磁力搅拌子的圆底烧瓶中,加入2-氯-3-羟基-6-碘吡啶2(789.2mg,3.09mmol)、溴化苄(793.4mg,4.64mmol)、k2co3(1281.1mg,9.27mmol,3.0equivs)和15.0ml四氢呋喃,在65℃下回流搅拌反应4小时,减压浓缩得到粗品,硅胶柱层析,洗脱剂:石油醚/乙酸乙酯=15:1,得白色固体2-氯-3-苄氧基-6-碘吡啶908.9mg,产率为85%;该过程如下反应:
[0085][0086]
化合物3的结构表征数据为:熔点82.8~84.4℃;1h nmr(400mhz,cdcl3)δ:7.37(d,j=8.3hz,1h),7.32
–
7.16(m,5h),6.79(d,j=8.3hz,1h),5.00(s,2h).
13
c nmr(100mhz,cdcl3)δ:151.1,140.6,135.0,134.0,128.8,128.5,127.1,123.0,102.0,71.1;hrms(esi)calcd.for c
12h10
clino[m h]
345.9490,found:345.9492。
[0087]
步骤2合成化合物4
[0088]
氩气保护下,在配有磁力搅拌的子的schlenk反应管中,依次加入化合物3(908.9mg,2.63mmol)、cs2co3(1427.1mg,4.38mmol,2.0equivs)、(
±
)-八角枫碱(355.2mg,2.19mmol)、正丁基二(1-金刚烷基)膦(62.8mg,8mol%)和催化剂pd(pph3)4(50.6mg,2mol%)和21.0ml 1,4-二氧六烷的溶液,在120℃下搅拌反应72小时,加入水淬灭反应,乙酸乙酯萃取3次,合并有机相,无水硫酸钠干燥,硅藻土过滤,所得溶液减压干燥,硅胶柱层析,洗脱剂:石油醚/乙酸乙酯=3:1,得无定形固体物,3-(benzyloxy)-2-chloro-6-(2-(pyridin-3-yl)pi peridin-1-yl)pyridine 444.5mg,产率为54%;该过程如下反应:
[0089][0090]
化合物4的结构表征数据为:1h nmr(400mhz,cdcl3)δ:8.43(d,j=2.4hz,1h),8.40
–
8.34(m,1h),7.48(dt,j=8.1,2.0hz,1h),7.38
–
7.12(m,6h),7.03(d,j=8.8hz,1h),6.32(d,j=8.8hz,1h),5.44(t,j=4.5hz,1h),4.94(s,2h),3.89(dq,j=13.7,5.1,4.2hz,1h),3.07(ddd,j=13.8,10.5,3.8hz,1h),2.13(dq,j=12.2,3.8hz,1h),1.94(dddd,j=14.0,11.6,6.0,3.6hz,1h),1.59(ddt,j=21.1,10.8,4.2hz,3h),1.49
–
1.35(m,1h);
13
c nmr(100mhz,cdcl3)δ:153.8,148.6,147.4,142.3,139.4,137.4,136.7,135.4,128.7,128.2,127.9,127.6,123.5,105.9,73.0,53.5,42.8,29.5,24.9,19.8;hrms(esi)calcd.for c
22h23
cln3o[m h]
380.1524,found:380.1517。
[0091]
步骤3合成化合物5
[0092]
氩气保护下,在配有磁力搅拌的子的schlenk反应管中,依次加入化合物4
(444.5mg,1.17mmol)、溶解于3-吡啶-苯硼酸(216.3mg,1.76mmol)、cs2co3(801.5mg,2.46mmol,2.1equivs)、k3po4(496.8mg,2.34mmol,2.0equivs)、催化剂pdcl2(pph3)2(4.1mg,5mol%),加入6.0ml甲苯和0.6ml乙醇,在100℃下搅拌反应139时,减压浓缩得到粗品,硅胶柱层析,洗脱剂:石油醚/乙酸乙酯=1:3,得无定形固体物,3-(benzyloxy)-6-(2-(pyridin-3-yl)piperidin-1-yl)-2,3'-bipyridine 350.8mg,产率为71%;该过程如下反应:
[0093][0094]
化合物5的结构表征数据为:1h nmr(400mhz,cdcl3)δ:9.13
–
9.08(m,1h),8.50
–
8.42(m,2h),8.37(d,j=4.8hz,1h),8.18(dt,j=8.0,1.9hz,1h),7.56(d,j=8.0hz,1h),7.31
–
7.14(m,8h),6.56(d,j=9.0hz,1h),5.53(t,j=4.8hz,1h),4.92(d,j=1.8hz,2h),3.91(dt,j=13.2,4.4hz,1h),3.20(ddd,j=13.6,10.0,4.1hz,1h),2.12(dq,j=13.6,4.4hz,1h),1.99(tdd,j=14.1,5.9,4.0hz,1h),1.74
–
1.42(m,4h);
13
c nmr(100mhz,cdcl3)δ:154.0,149.8,148.5,147.9,147.1,145.9,141.7,138.4,137.1,136.6,135.4,134.1,128.6,128.1,127.4,126.2,123.5,123.0,108.4,72.3,53.8,43.8,30.1,25.0,20.1;hrms(esi)calcd.for c
27h27
n4o[m h]
423.2179,found:423.2175。
[0095]
步骤4合成化合物1
[0096]
氩气保护下,在配有磁力搅拌的子的schlenk反应管中,依次加入溶解nah(60%in oil,49.8mg,1.5equivs)和pd(oac)2(9.3mg,5mol%)和4.0ml二甲基乙酰胺,在室温下搅拌5分钟后,加入化合物5(350.8mg,0.83mmol)的二甲基乙酰胺溶液,再将该混合物在60℃下搅拌反应3小时。随后加入10.0ml饱和氯化铵水溶液,再用乙酸乙酯萃取(10.0ml
×
3次),合并有机相,用水反萃(10.0ml
×
3次),有机相用无水硫酸钠干燥,硅藻土过滤,减压浓缩得到粗品,硅胶柱层析,洗脱剂:二氯甲烷/甲醇/氨水=250:26:1,得无定形固体物,6-(2-(pyridin-3-yl)piperidin-1-yl)-[2,3'-bipyridin]-3-ol 273.1mg,产率为99%;该过程如下反应:
[0097][0098]
化合物1的结构表征数据为:1h nmr(400mhz,cd3od,图9)δ:9.14
–
9.09(m,1h),8.46(d,j=2.3hz,1h),8.43
–
8.35(m,2h),8.30(dd,j=4.9,1.6hz,1h),7.79(dt,j=8.0,2.0hz,1h),7.42(ddd,j=8.1,5.0,0.9hz,1h),7.33(dd,j=8.0,4.9hz,1h),7.19(d,j=8.9hz,1h),6.82(d,j=8.9hz,1h),5.31(t,j=5.4hz,1h),3.69(ddd,j=12.8,6.0,4.4hz,1h),3.38(ddd,j=12.6,8.1,4.3hz,1h),2.06(q,j=5.7hz,2h),1.79(qt,j=8.8,5.0hz,2h),1.70
–
1.56(m,2h);
13
c nmr(100mhz,cd3od,图10)δ:155.2,150.2,149.2,148.1,147.6,146.7,141.8,138.5,138.1,137.2,136.3,128.5,125.0,124.5,113.3,57.0,48.1,32.9,
26.5,22.4.hrms(esi,图8)calcd.for c
20h21
n4o[m h]
333.1710,found:333.1713。与制备的天然产物(
±
)-alanginenmine a的核磁数据基本一致。
[0099]
实施例5
[0100]
取(
±
)-alanginenmine a化合物作为原料药,加入1/9的糊精,制粒,得颗粒剂。
[0101]
实施例6
[0102]
取(
±
)-alanginenmine a化合物作为原料药,加入1/9的糊精,混匀,装入胶囊,得胶囊剂。
[0103]
实施例7
[0104]
取(
±
)-alanginenmine a化合物作为原料药,加入1/10的糊精混合均匀,干燥,制成丸剂。
[0105]
实施例8
[0106]
取(
±
)-alanginenmine a化合物作为原料药,加入1/10的糊精,制粒,压片,制成片剂。
[0107]
实施例9
[0108]
取(
±
)-alanginenmine a化合物作为原料药,加入13倍量的注射用水,搅拌,过滤,灭菌,得注射剂。
[0109]
实施例10
[0110]
取(
±–
)-alanginenmine a化合物作为原料药,加入9倍量的注射用水,搅拌,过滤,冻干,得冻干粉剂。
[0111]
实施例11
[0112]
取(
±
)-alanginenmine a化合物作为原料药,加入13倍量纯净水,混合均匀,过滤,灭菌,得口服液。
[0113]
应用:(
±
)-alanginenmine a在制备抗hiv药物或药物制剂中的应用。
[0114]
实验例1
[0115]
哌啶类生物碱的提取纯化方法:
[0116]
取5kg干燥的八角枫alangium chinense(lour.)harms根,粉碎后用甲醇回流提取3次,每次20l,每次3小时,得到甲醇提取物。减压除去有机溶剂后,得到醇提浸膏461g,边搅拌边加入hcl溶液将ph调至2,提取物溶解后过滤。再用氨水将滤液的ph值调节至10,然后用5l的etoac萃取4次,得到萃取液,减压浓缩得浸膏40g。将该浸膏进行硅胶柱层析,用1:0~0:1的二氯甲烷-甲醇溶剂梯度洗脱,得到7个组分a-g,其中组分b为20g。组分b进行硅胶柱层析,以用1:0~0:1的石油醚-乙酸乙酯溶剂梯度洗脱,得到11个组分(b1~b
11
),其中b
10
为639mg。b
10
进行sephadex lh-20柱色谱分离,以甲醇做洗脱剂,得到含化合物1的粗品130mg,随后该粗品用mci-gel chp 20p柱分离,以1:4~4:1的甲醇-水作为洗脱剂,梯度洗脱,得到6.69mg的化合物1。
[0117]
化合物表征:
[0118]
化合物1:(
±
)-alanginenmine a
[0119]
结构式:
[0120][0121]
分子式:c
20h20
n4o
[0122]
状态:无定形固体
[0123]
hresims m/z:333.1707[m h]
[0124]
硅胶薄层检识:以比例为110:10:1的二氯甲烷:乙酸乙酯:氨水展开(rf值为0.5),在254nm下有荧光,经碘化铋钾显色剂显色后呈现红色斑点,经茚三酮显色烘烤呈浅紫色斑点;
[0125]1h-nmr(600mhz,cd3od)、
13
c-nmr(150mhz,cd3od)数据如下:
[0126][0127]
实验例2
[0128]
半合成制备(
±
)-alanginenmine a:
[0129]
步骤1合成化合物3
[0130]
在配备有磁力搅拌子的圆底烧瓶中,加入2-氯-3-羟基-6-碘吡啶2(789.2mg,3.09mmol)、溴化苄(793.4mg,4.64mmol)、k2co3(1281.1mg,9.27mmol,3.0equivs)和15.0ml
四氢呋喃,在65℃下回流搅拌反应4小时,减压浓缩得到粗品,硅胶柱层析,洗脱剂:石油醚/乙酸乙酯=15:1,得白色固体2-氯-3-苄氧基-6-碘吡啶908.9mg,产率为85%;该过程如下反应:
[0131][0132]
化合物3的结构表征数据为:熔点82.8~84.4℃;1h nmr(400mhz,cdcl3)δ:7.37(d,j=8.3hz,1h),7.32
–
7.16(m,5h),6.79(d,j=8.3hz,1h),5.00(s,2h).
13
c nmr(100mhz,cdcl3)δ:151.1,140.6,135.0,134.0,128.8,128.5,127.1,123.0,102.0,71.1;hrms(esi)calcd.for c
12h10
clino[m h]
345.9490,found:345.9492。
[0133]
步骤2合成化合物4
[0134]
氩气保护下,在配有磁力搅拌的子的schlenk反应管中,依次加入化合物3(908.9mg,2.63mmol)、cs2co3(1427.1mg,4.38mmol,2.0equivs)、(
±
)-八角枫碱(355.2mg,2.19mmol)、正丁基二(1-金刚烷基)膦(62.8mg,8mol%)和催化剂pd(pph3)4(50.6mg,2mol%)和21.0ml 1,4-二氧六烷的溶液,在120℃下搅拌反应72小时,加入水淬灭反应,乙酸乙酯萃取3次,合并有机相,无水硫酸钠干燥,硅藻土过滤,所得溶液减压干燥,硅胶柱层析,洗脱剂:石油醚/乙酸乙酯=3:1,得无定形固体物,3-(benzyloxy)-2-chloro-6-(2-(pyridin-3-yl)pi peridin-1-yl)pyridine 444.5mg,产率为54%;该过程如下反应:
[0135][0136]
化合物4的结构表征数据为:1h nmr(400mhz,cdcl3)δ:8.43(d,j=2.4hz,1h),8.40
–
8.34(m,1h),7.48(dt,j=8.1,2.0hz,1h),7.38
–
7.12(m,6h),7.03(d,j=8.8hz,1h),6.32(d,j=8.8hz,1h),5.44(t,j=4.5hz,1h),4.94(s,2h),3.89(dq,j=13.7,5.1,4.2hz,1h),3.07(ddd,j=13.8,10.5,3.8hz,1h),2.13(dq,j=12.2,3.8hz,1h),1.94(dddd,j=14.0,11.6,6.0,3.6hz,1h),1.59(ddt,j=21.1,10.8,4.2hz,3h),1.49
–
1.35(m,1h);
13
c nmr(100mhz,cdcl3)δ:153.8,148.6,147.4,142.3,139.4,137.4,136.7,135.4,128.7,128.2,127.9,127.6,123.5,105.9,73.0,53.5,42.8,29.5,24.9,19.8;hrms(esi)calcd.for c
22h23
cln3o[m h]
380.1524,found:380.1517。
[0137]
步骤3合成化合物5
[0138]
氩气保护下,在配有磁力搅拌的子的schlenk反应管中,依次加入化合物4(444.5mg,1.17mmol)、溶解于3-吡啶-苯硼酸(216.3mg,1.76mmol)、cs2co3(801.5mg,2.46mmo l,2.1equivs)、k3po4(496.8mg,2.34mmol,2.0equivs)、催化剂pdcl2(pph3)2(4.1mg,5mol%),加入6.0ml甲苯和0.6ml乙醇,在100℃下搅拌反应139时,减压浓缩得到粗品,硅胶柱层析,洗脱剂:石油醚/乙酸乙酯=1:3,得无定形固体物,3-(benzyloxy)-6-(2-(p yridin-3-yl)piperidin-1-yl)-2,3'-bipyridine 350.8mg,产率为71%;该过程如下
反应:
[0139][0140]
化合物5的结构表征数据为:1h nmr(400mhz,cdcl3)δ:9.13
–
9.08(m,1h),8.50
–
8.42(m,2h),8.37(d,j=4.8hz,1h),8.18(dt,j=8.0,1.9hz,1h),7.56(d,j=8.0hz,1h),7.31
–
7.14(m,8h),6.56(d,j=9.0hz,1h),5.53(t,j=4.8hz,1h),4.92(d,j=1.8hz,2h),3.91(dt,j=13.2,4.4hz,1h),3.20(ddd,j=13.6,10.0,4.1hz,1h),2.12(dq,j=13.6,4.4hz,1h),1.99(tdd,j=14.1,5.9,4.0hz,1h),1.74
–
1.42(m,4h);
13
c nmr(100mhz,cdcl3)δ:154.0,149.8,148.5,147.9,147.1,145.9,141.7,138.4,137.1,136.6,135.4,134.1,128.6,128.1,127.4,126.2,123.5,123.0,108.4,72.3,53.8,43.8,30.1,25.0,20.1;hrms(esi)calcd.for c
27h27
n4o[m h]
423.2179,found:423.2175。
[0141]
步骤4合成化合物1
[0142]
氩气保护下,在配有磁力搅拌的子的schlenk反应管中,依次加入溶解nah(60%in oil,49.8mg,1.5equivs)和pd(oac)2(9.3mg,5mol%)和4.0ml二甲基乙酰胺,在室温下搅拌5分钟后,加入化合物5(350.8mg,0.83mmol)的二甲基乙酰胺溶液,再将该混合物在60℃下搅拌反应3小时。随后加入10.0ml饱和氯化铵水溶液,再用乙酸乙酯萃取(10.0ml
×
3次),合并有机相,用水反萃(10.0ml
×
3次),有机相用无水硫酸钠干燥,硅藻土过滤,减压浓缩得到粗品,硅胶柱层析,洗脱剂:二氯甲烷/甲醇/氨水=250:26:1,得无定形固体物,6-(2-(pyridin-3-yl)piperidin-1-yl)-[2,3'-bipyridin]-3-ol 273.1mg,产率为99%;该过程如下反应:
[0143][0144]
化合物1的结构表征数据为:1h nmr(400mhz,cd3od)δ:9.14
–
9.09(m,1h),8.46(d,j=2.3hz,1h),8.43
–
8.35(m,2h),8.30(dd,j=4.9,1.6hz,1h),7.79(dt,j=8.0,2.0hz,1h),7.42(ddd,j=8.1,5.0,0.9hz,1h),7.33(dd,j=8.0,4.9hz,1h),7.19(d,j=8.9hz,1h),6.82(d,j=8.9hz,1h),5.31(t,j=5.4hz,1h),3.69(ddd,j=12.8,6.0,4.4hz,1h),3.38(ddd,j=12.6,8.1,4.3hz,1h),2.06(q,j=5.7hz,2h),1.79(qt,j=8.8,5.0hz,2h),1.70
–
1.56(m,2h);
13
c nmr(100mhz,cd3od)δ:155.2,150.2,149.2,148.1,147.6,146.7,141.8,138.5,138.1,137.2,136.3,128.5,125.0,124.5,113.3,57.0,48.1,32.9,26.5,22.4.hrms(esi)calcd.for c
20h21
n4o[m h]
333.1710,found:333.1713。与制备的天然产物(
±
)-alanginenmine a的核磁数据基本一致。
[0145]
本发明的化合物1具有潜在的抗hiv生物活性,体外抗hiv活性测定试验表明:本发明的化合物1具有潜在的重要生物活性,可以作为抗hiv病毒的潜在抑制剂。
[0146]
实验例3:药理实验:体外抗hiv活性测定
[0147]
使用sensolyte 520hiv pr检测试剂盒,测定单体化合物对hiv-1蛋白酶的抑制活性。体外抗hiv蛋白酶活性测定结果显示,化合物1显示出对hiv-1蛋白酶的抑制作用,化合物1在0.1mg/ml、0.01mg/ml和0.001mg/ml(dmso)的给药浓度下,抑制率分别为100%、45.2%和24.1%,化合物1的ic
50
值为15.1μmol/l,见表1。
[0148]
表1化合物1对hiv-1蛋白酶的抑制活性
[0149][0150]a阳性对照药
[0151]
虽然,上文中已经用一般性说明、具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作出一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。