1.本发明涉及一种西他列汀的合成方法,属于药物制备领域。
背景技术:
2.西他列汀(sitagliptin),由美国默克公司研制开发,于治疗ii型糖尿病的二肽基肽酶-iv抑制剂药物,通过提高糖尿病患者自身胰岛β细胞产生胰岛素的能力,在血糖升高时增加患者体内胰岛素的分泌,从而控制糖尿病患者的血糖水平,不仅治疗效果好,而且不会产生耐受性,在治疗的过程中也不会带来低血糖的风险。
3.其活性成分为(3r)-3-氨基-1-[3-(三氟甲基)-5,6-二氢-1,2,4-三唑并[4,3-a]吡嗪-7(8h)-基]-4-(2,4,5-三氟苯基)丁-1-酮(式一),其结构式如下所示:
[0004][0005]
目前西他列汀的合成方法较多,涉及手性诱导及不对称氢化的步骤少则四五步,多则十多步,且多数方法合成路线长,工序繁琐,而且反应过程中使用的催化剂价格昂贵,不利于大规模工业生产。
[0006]
如在cn102153559b中,采用l-天冬氨酸改造后作为原料与2,4,5-三氟卤苯反应的例子,但因为要先将三氟卤苯做成锌试剂,所以反应过程中必须使用昂贵的钯催化剂对反应进行催化,生产成本高不利于生产放大,该合成工艺的路线如下所示:
[0007][0008]
专利wo03004498中报道了一种由酯类中间物经过贵金属和催化得到中间体的合成路线,反应过程中用到了贵金属铑以及二茂铁基双磷,都是价格昂贵的物质,不适合工业化的放大生产。
[0009]
专利wo2010078440报道了2,4,5-三氟苯乙酸经与丙二酸镁盐经缩合得到酯类中间物的合成路线,该方法的原料同样较为昂贵,增加了生产成本,限制了工业化生产,该合
成工艺的路线如下所示:
[0010][0011]
从以上文献中可以看出其共同存在的缺点是生产成本较高,限制了工业化生产。
[0012]
因此,目前亟需寻找一种生产成本低、反应路线短、产物率高的合成方法。
技术实现要素:
[0013]
本发明针对以上现有技术中存在的缺陷,提供一种西他列汀的合成方法,解决的问题是如何实现减少生产成本、缩短反应路线的制备方法。
[0014]
本发明的目的是通过以下技术方案得以实现的,一种西他列汀的合成方法,该方法包括以下步骤:
[0015]
s1:以l-天冬氨酸-4-酯为起始原料,生成中间化合物(s)-4-对甲苯磺酰氧基-3-叔丁氧羰基氨基丁酸甲酯;
[0016]
s2:将s1步骤中获得的中间化合物与卤化物反应生成式三化合物;
[0017]
s3:将s2步骤中得到的化合物与2,4,5三氟苯甲醛反应生成中间化合物(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸甲酯;
[0018]
s4:将s3步骤中得到的中间化合物水解得到式二化合物西他列汀中间体(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸;
[0019]
s5:将西他列汀中间体(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸与3-(三氟甲基)-5,6,7,8-四氢-[1,2,4]三唑并[4,3-a]吡嗪盐酸盐缩合得到西他列汀的叔丁基氧羰基缩醛;
[0020]
s6:将s5步骤中缩醛产物化合物经盐酸甲醇脱保护得到十一化合物西他列汀。
[0021]
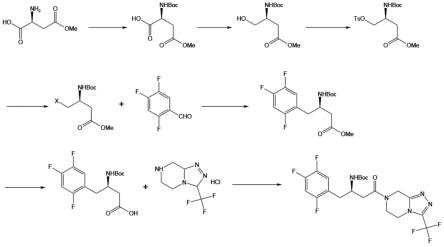
总合成路线为:
[0022]
[0023]
式一化合物合成路线为:
[0024][0025]
式三化合物合成路线:
[0026][0027]
式二化合物合成路线:
[0028][0029]
本发明通过采用l-天冬氨酸-4-酯改造后经过反应得到的最终产物卤代叔丁氧羰基氨基丁酸甲酯保留了l-天冬氨酸的手性氨基结构,再与2,4,5-三氟苯甲醛反应合成中间化合物后只需要通过水解即可得到西他列汀中间体(式二),再将得到的式二化合物与3-(三氟甲基)-5,6,7,8-四氢-[1,2,4]三唑并[4,3-α]吡嗪盐酸盐进行缩合即可生成西他列汀的叔丁基氧羰基缩醛,经盐酸甲醇脱保护就能得到西他列汀,由于使用了l-天冬氨酸作为反应原料保留了手性氨基结构,因此省去了单独合成手性氨基结构的复杂过程,并且反应生成的中间化合物能够直接与2,4,5-三氟苯甲醛反应生成西他列汀中间体,使用的催化剂便宜易得,无需和现有技术一样使用昂贵的钯试剂或者贵金属催化,减少了生产成本的同时还能降低后处理难度以及反应过程会发生的操作风险。
[0030]
式一化合物结构式如下:
[0031][0032]
式二化合物结构式如下:
[0033][0034]
式三化合物结构式如下:
[0035][0036]
其中,式三化合物中x为溴、氯或碘。
[0037]
在上述西他列汀中间体的合成方法中,作为优选,s1步骤中的溶剂可选用二氯甲烷或二氯乙烷其中一种,能够更好的溶解中间化合物。作为优选,可选用二氯甲烷作为溶液,价格便宜的同时反应速率更快。
[0038]
在上述西他列汀中间体的合成方法中,作为优选,s2步骤中使用的试剂为溴化钠、氯化钾与碘化钠其中一种,生成能够与苯甲醛反应的中间化合物。作为优选,选择溴化钠作为催化剂,相比较反应速率更快,反应更加彻底。
[0039]
在上述西他列汀中间体的合成方法中,作为优选,s2步骤中卤代反应温度为68℃~80℃,能够使反应速率得到提升。作为优选,反应温度可设为75℃,能够缩短反应时间。
[0040]
在上述西他列汀中间体的合成方法中,作为优选,s3步骤反应发生在醚类溶剂中,醚类溶剂可选用四氢呋喃、二氧六环中的一种。采用醚类溶剂易于操作,对环境污染相对较
少,能够减少整体的生产成本点。作为优选,所述醚类溶剂可选用四氢呋喃,能够提高反应稳定性,更好的促进反应的进行,提高产物收率。
[0041]
在上述西他列汀中间体的合成方法中,作为优选,s4步骤中反应温度为-70℃~-80℃。低温下反应过程放缓,反应更加稳定。作为优选,反应温度可设为-78℃,增加反应化合物与反应溶剂的稳定性,便于反应进行。
[0042]
在上述西他列汀中间体的合成方法中,作为优选,s5步骤在醇类溶剂中发生,可选用甲醇、乙醇或丙醇的其中一种。醇溶剂有着更好的包容性和稳定性,能够使反应过程稳定进行,同时价格便宜,有利于降低生产成本。作为优选,可选用甲醇作为反应溶剂,使反应过程更加稳定。
[0043]
在上述西他列汀中间体的合成方法中,作为优选,s6步骤中使用的催化剂可选用二异丙基乙基胺与三乙胺其中一种,能够提升反应效率。作为优选,选用二异丙基乙基胺的副产物更加易于处理。
[0044]
综上所述,本发明与现有技术相比,具有以下优点:
[0045]
1.本发明的反应路线使用的催化剂价格低廉,相比较其他反应路线中所使用的昂贵催化剂如钯催化剂与铑催化剂以及二茂铁基双磷,能够减少生产成本,利于扩大生产。
[0046]
2.l-天冬氨酸-4-酯作为起始原料生成的产物保留了l-天冬氨酸的手性氨基结构,减少了后期合成手性氨基的复杂步骤,并且得到的中间化合物只需要水解就能生成西他列汀中间体,进一步提升了反应效率。
附图说明
[0047]
图1为本发明总合成路线;
[0048]
图2为本发明式三化合物合成路线;
[0049]
图3为本发明式二化合物合成路线;
[0050]
图4为本发明式一化合物合成路线;
[0051]
图5为式一化合物结构式;
[0052]
图6为式二化合物结构式;
[0053]
图7为式三化合物结构式。
具体实施方式
[0054]
下面通过具体实施例,对本发明的技术方案作进一步具体的说明,但是本发明并不限于这些实施例。
[0055]
实施例1
[0056]
第一步:n-boc-l-天冬氨酸-4-甲酯的制备:
[0057]
将l-天冬氨酸-4-甲酯盐酸盐91.8g溶解于1000ml水中,然后缓慢加入碳酸氢钠42.0g,加完后搅拌30分钟。再滴加含有boc酸酐109.1g的丙酮溶液750ml,滴加完毕后室温搅拌过夜。反应结束后,过滤,少量丙酮洗滤饼,合并滤液后蒸除丙酮,用500ml乙酸乙酯萃取水相,合并的有机相用饱和食盐水洗、无水硫酸钠干燥1小时、过滤、浓缩得到无色油状物n-boc-l-天冬氨酸-4-甲酯105.9g,收率为85.7%。
[0058]
第二步:(s)-4-羟基-3-叔丁氧羰基氨基丁酸甲酯的制备
[0059]
在氮气的保护下,n-boc-l-天冬氨酸-4-甲酯123.6g溶解于1000ml四氢呋喃中,反应温度降至-10℃,加入n-甲基吗啉50.5g。控温-10℃,开始滴加氯甲酸乙酯54.2g,滴加完毕后搅拌10分钟。分批加入硼氢化钠共18.9g,加完后缓慢滴入1000ml甲醇,控制气体产生的速度,并控温-3℃以下,滴加完毕后继续搅拌30min,蒸除四氢呋喃、甲醇,加100ml水溶解,再用乙酸乙酯萃取(750ml
×
3),合并有机相,分别用硫酸氢钾、5%的碳酸氢钠以及饱和食盐水洗涤,无水硫酸钠干燥1小时,过滤、得到浅色油状产物(s)-4-羟基-3-叔丁氧羰基氨基丁酸甲酯76.9g,收率为66%。
[0060]
第三步:(s)-4-对甲苯磺酰氧基-3-叔丁氧羰基氨基丁酸甲酯的制备
[0061]
在氮气的保护下,将(s)-4-羟基-3-叔丁氧羰基氨基丁酸甲酯116.6g溶于500ml二氯甲烷中,冰水浴冷至0℃,加入50.6g三乙胺,然后再滴加含有36.57g对甲苯磺酰氯的二氯甲烷溶液200ml,滴加完毕后,升温至室温。反应结束后,加100ml水分出有机相,水相用少量二氯甲烷萃取,使用饱和碳酸氢钠洗涤有机相,无水硫酸钠干燥1小时、过滤、得到浅色油状产物(s)-4-对甲苯磺酰氧基-3-叔丁氧羰基氨基丁酸甲酯111.58g,收率为57.6%。
[0062]
第四步:(s)-4-溴-3-叔丁氧羰基氨基丁酸甲酯(式三)的制备
[0063]
在氮气保护下,将(s)-4-对甲苯磺酰氧基-3-叔丁氧羰基氨基丁酸甲酯193.7g溶入1000ml丙酮中,加入溴化钠51.4g,然后再加入溴化铁147.7g,升温至75℃,搅拌12小时,反应结束后,减压蒸去部分溶剂,再往反应体系中加入5%的亚硫酸氢钠溶液200ml,体系用100ml乙酸乙酯萃取2次,合并有机相用水洗两次,饱和食盐水洗一次,无水硫酸钠干燥2h,过滤,减压蒸去溶剂,得到淡黄色油状液体(s)-4-溴-3-叔丁氧羰基氨基丁酸甲酯164.7g,收率为96%。
[0064]
第五步:(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸(式二)的制备
[0065]
在氮气保护下,将(s)-4-溴-3-叔丁氧羰基氨基丁酸甲酯171.6g溶于500ml四氢呋喃中,加入二甲硫醚溴化亚铜20.5g,使之分散均匀。反应体系降温至-78℃,加入88.27g的2,4,5-三氟苯甲醛与500ml四氢呋喃溶液,维持温度-78℃左右,保温30min,缓慢升至室温,搅拌过夜,反应完成后,加100ml水搅拌1小时,加入50ml乙酸乙酯分层,无水硫酸钠干燥1h,得到(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸甲酯143.1g,收率为82.4%。
[0066]
将(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸甲酯173.6g溶解于500ml甲醇中,搅拌加入12g氢氧化锂的100ml水溶液,室温反应2-3小时,反应结束后,缓慢加入硫酸氢钠的水溶液,ph调至3后停止,加入50ml乙酸乙酯萃取2次,合并的有机相用无水硫酸钠干燥,浓缩,得白色固体产物(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸160.8g,收率为96.5%。
[0067]
实施例2
[0068]
本实施例为西他列汀的制备
[0069]
将166.7g的(r)-3-((r)-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸和114.3g的3-(三氟甲基)-5,6,7,8-四氢-[1,2,4]三唑并[4,3-a]吡嗪盐酸盐溶于1000ml二氯甲烷溶液中,降温至5℃,再缓慢滴加二异丙基乙基胺64.62g,滴毕,保温搅拌30min,将67.5g的1-羟基苯并三氮唑分批加入溶液中,控温5℃继续搅拌60min,再次分批加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐95.9g,室温搅拌过夜,得到中间产物n-叔丁氧羰基-西他列汀227.58g,收率为89.7%。
[0070]
室温下将253.7gn-叔丁氧羰基-西他列汀加入1000ml的hcl/meoh溶液中,室温搅拌12小时,反应结束后将反应液倒入1000ml水中,加入100ml乙酸乙酯萃取水相,干燥后浓缩后得到182.06g最终产物西他列汀,收率为89.4%。
[0071]
实施例3
[0072]
在氮气的保护下,将(s)-4-羟基-3-叔丁氧羰基氨基丁酸甲酯116.6g溶于500ml二氯乙烷中,冰水浴冷至0℃,加入50.6g三乙胺,然后再滴加含有36.57g对甲苯磺酰氯的二氯乙烷溶液200ml,滴加完毕后,升温至室温。反应结束后,加100ml水分出有机相,水相用少量二氯乙烷萃取,使用饱和碳酸氢钠洗涤有机相,无水硫酸钠干燥1小时、过滤、得到浅色油状产物(s)-4-对甲苯磺酰氧基-3-叔丁氧羰基氨基丁酸甲酯100.73g,收率为52%。
[0073]
实施例4
[0074]
在氮气保护下,将(s)-4-对甲苯磺酰氧基-3-叔丁氧羰基氨基丁酸甲酯193.7g溶入1000ml丙酮中,加入氯化钾37.27g,然后再加入溴化铁147.7g,升温至75℃,搅拌12小时,反应结束后,减压蒸去部分溶剂,再往反应体系中加入5%的亚硫酸氢钠溶液200ml,体系用100ml乙酸乙酯萃取2次,合并有机相用水洗两次,饱和食盐水洗一次,无水硫酸钠干燥2h,过滤,减压蒸去溶剂,得到淡黄色油状液体(s)-4-氯-3-叔丁氧羰基氨基丁酸甲酯146.68g,收率为85.5%。
[0075]
实施例5
[0076]
在氮气保护下,将(s)-4-对甲苯磺酰氧基-3-叔丁氧羰基氨基丁酸甲酯193.7g溶入1000ml丙酮中,加入碘化钠74.9g,然后再加入溴化铁147.7g,升温75℃,搅拌12小时,反应结束后,减压蒸去部分溶剂,再往反应体系中加入5%的亚硫酸氢钠溶液200ml,体系用100ml乙酸乙酯萃取2次,合并有机相用水洗两次,饱和食盐水洗一次,无水硫酸钠干燥2h,过滤,减压蒸去溶剂,得到淡黄色油状液体(s)-4-碘-3-叔丁氧羰基氨基丁酸甲酯134.1g,收率为78.2%。
[0077]
实施例6
[0078]
在氮气保护下,将(s)-4-对甲苯磺酰氧基-3-叔丁氧羰基氨基丁酸甲酯193.7g溶入1000ml丙酮中,加入溴化钠51.4g,然后再加入溴化铁147.7g,升温至68℃,搅拌12小时,反应结束后,减压蒸去部分溶剂,再往反应体系中加入5%的亚硫酸氢钠溶液200ml,体系用100ml乙酸乙酯萃取2次,合并有机相用水洗两次,饱和食盐水洗一次,无水硫酸钠干燥2h,过滤,减压蒸去溶剂,得到淡黄色油状液体(s)-4-溴-3-叔丁氧羰基氨基丁酸甲酯152.8g,收率为89.1%
[0079]
实施例7
[0080]
在氮气保护下,将(s)-4-对甲苯磺酰氧基-3-叔丁氧羰基氨基丁酸甲酯193.7g溶入1000ml丙酮中,加入溴化钠51.4g,然后再加入溴化铁147.7g,升温至80℃,搅拌12小时,反应结束后,减压蒸去部分溶剂,再往反应体系中加入5%的亚硫酸氢钠溶液200ml,体系用100ml乙酸乙酯萃取2次,合并有机相用水洗两次,饱和食盐水洗一次,无水硫酸钠干燥2h,过滤,减压蒸去溶剂,得到淡黄色油状液体(s)-4-溴-3-叔丁氧羰基氨基丁酸甲酯156.8g,收率为91.4%
[0081]
实施例8
[0082]
在氮气保护下,将(s)-4-溴-3-叔丁氧羰基氨基丁酸甲酯171.6g溶于500ml二氧六
环中,加入二甲硫醚溴化亚铜20.5g,使之分散均匀。反应体系降温至-78℃,加入88.27g的2,4,5-三氟苯甲醛与500ml二氧六环溶液,维持温度-78℃左右,保温30min,缓慢升至室温,搅拌过夜,反应完成后,加100ml水搅拌1小时,加入50ml乙酸乙酯分层,无水硫酸钠干燥1h,得到(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸甲酯133.02g,收率为76.6%。
[0083]
实施例9
[0084]
在氮气保护下,将(s)-4-溴-3-叔丁氧羰基氨基丁酸甲酯171.6g溶于500ml四氢呋喃中,加入二甲硫醚溴化亚铜20.5g,使之分散均匀。反应体系降温至-80℃,加入88.27g的2,4,5-三氟苯甲醛与500ml四氢呋喃溶液,维持温度-80℃左右,保温30min,缓慢升至室温,搅拌过夜,反应完成后,加100ml水搅拌1小时,加入50ml乙酸乙酯分层,无水硫酸钠干燥1h,得到(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸甲酯131.46g,收率为75.7%。
[0085]
实施例10
[0086]
在氮气保护下,将(s)-4-溴-3-叔丁氧羰基氨基丁酸甲酯171.6g溶于500ml四氢呋喃中,加入二甲硫醚溴化亚铜20.5g,使之分散均匀。反应体系降温至-70℃,加入88.27g的2,4,5-三氟苯甲醛与500ml四氢呋喃溶液,维持温度-70℃左右,保温30min,缓慢升至室温,搅拌过夜,反应完成后,加100ml水搅拌1小时,加入50ml乙酸乙酯分层,无水硫酸钠干燥1h,得到(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸甲酯125.7g,收率为72.4%。
[0087]
实施例11
[0088]
降温至5℃,将186.4g甲氧基甲基三苯基氯化膦溶于500ml二氧六环中,分批加入加24g氢化钠完后升至室温搅拌6小时,随后加入2,4,5-三氟苯甲醛80.5g,室温搅拌12小时,抽滤,得到淡黄色油状物1,2,4-三氟-5-(2-甲氧乙烯基)苯80.7g,收率为85.8%。
[0089]
将94g的1,2,4-三氟-5-(2-甲氧乙烯基)苯溶于500ml二氧六环溶液中,降温至-20-20℃,缓慢滴加20ml浓盐酸,加完后升温至40℃搅拌3小时,将反应液倒入水中,加入50ml乙酸乙酯萃取水相,用饱和食盐水盐水洗涤有机相,抽去溶剂,浓缩后得到75.75g淡黄色油状物2-(2,4,5-三氟苯基)乙醛,收率为87%。
[0090]
实施例12
[0091]
将(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸甲酯173.6g溶解于500ml乙醇中,搅拌加入12g氢氧化锂的100ml水溶液,室温反应2-3小时,反应结束后,缓慢加入硫酸氢钠的水溶液,ph调至3后停止,加入50ml乙酸乙酯萃取2次,合并的有机相用无水硫酸钠干燥,浓缩,得白色固体产物(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸148.3g,收率为89%。
[0092]
实施例13
[0093]
将(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸甲酯173.6g溶解于500ml丙醇中,搅拌加入12g氢氧化锂的100ml水溶液,室温反应2-3小时,反应结束后,缓慢加入硫酸氢钠的水溶液,ph调至3后停止,加入50ml乙酸乙酯萃取2次,合并的有机相用无水硫酸钠干燥,浓缩,得白色固体产物(r)-3-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸145.9g,收率为87.6%。
[0094]
实施例14
[0095]
将166.7g的(r)-3-((r)-叔丁氧羰基氨基-4-(2,4,5-三氟苯基)丁酸和114.3g的3-(三氟甲基)-5,6,7,8-四氢-[1,2,4]三唑并[4,3-a]吡嗪盐酸盐溶于1000ml二氯甲烷溶
液中,降温至5℃,再缓慢滴加三乙胺50.6g,滴毕,保温搅拌30min,将67.5g的1-羟基苯并三氮唑分批加入溶液中,控温5℃继续搅拌60min,再次分批加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐95.9g,室温搅拌过夜,得到中间产物n-叔丁氧羰基-西他列汀210.3g,收率为82.9%。
[0096]
实施例15
[0097]
本实施例为cn102153559a公开文件的实施例
[0098]
第一步:氨基保护
[0099]
在装有磁力搅拌器,温度计的三口烧瓶中加入l-天冬氨酸20g,甲醇200ml,冷却滴加5ml氯化亚砜,滴加完室温搅拌5h,减压浓缩脱溶剂得到固体4-l-天冬氨酸甲酯盐酸盐40g。
[0100]
在装有磁力搅拌器,温度计的三口烧瓶中加入4-l-天冬氨酸甲酯盐酸盐20g,碳酸氢钠20g,溶于水和1,4-二氧六环中,溶清后加入boc酸酐,搅拌过夜,抽滤,母液加水2l,用盐酸调节ph。然后用乙酸乙酯萃取3次,无水硫酸钠干燥,减压浓缩脱溶剂得到淡黄色油状液体n-叔丁氧羰基-4-l-天冬氨酸甲酯31g。
[0101]
第二步:酯化
[0102]
将30g n-叔丁氧羰基-4-l-天冬氨酸甲酯溶于乙酸乙酯中,再加n-羟基丁二酰亚胺29g,搅拌滴加30g edci,溶于100ml乙酸乙酯,搅拌,抽滤,母液洗涤两次,有机相无水硫酸钠干燥,减压浓缩脱溶剂,得淡黄色油状液体(3s)-n-叔丁氧羰基-3-氨基-4-丁二酰亚胺氧基丁酸甲酯39g。
[0103]
第三步:还原和碘代
[0104]
还原:取1.4g硼氢化钠溶于100mlthf,滴加到20g(3s)-n-叔丁氧羰基-3-氨基-4-碘丁酸甲酯和50ml thf中,有大量气泡生成,温度升高至30℃,滴毕,点板,反应完全,乙酸乙酯萃取2次,无水硫酸钠干燥,减压浓缩脱溶剂得5.5g。
[0105]
碘代:9.8g三苯基磷溶于70ml二氯甲烷,溶清后加入1g咪唑,加2g碘,温度上升至34℃,反应20min后,慢慢加入3.8g上述得到的淡黄色油状液体,点板跟踪反应,原料点消失后抽滤,饱和食盐水洗2次,有机层无水硫酸钠干燥,减压蒸去二氯甲烷,得白色固体(3s)-n-叔丁氧羰基-3-氨基-4-碘丁酸甲酯3.5g。
[0106]
第四步:偶练
[0107]
取锌粉1.48g,2.5mldmf,50℃氮气保护下加入1,2-二溴乙烷1ml,搅拌反应20min,停止加热,30℃下加入三甲基氯硅烷,反应微弱放热,20min后加入1.3g(3s)-n-叔丁氧羰基-3-氨基-4-碘丁酸甲酯和10mldmf溶液,点板跟踪反应,30min后原料点消失,加催化剂双三苯基磷氯化钯0.2g,30℃下注射器慢慢注入2,4,5-三氟碘苯1.27g,反应放热,加入完毕,高效液相跟踪反应,原料点消失后乙酸乙酯50ml稀释反应液,抽滤,母液,饱和食盐水洗涤2次,水洗2次,无水硫酸钠干燥,减压浓缩得产品(3r)-n-叔丁氧羰基-3-氨基-4-(2,4,5-三氟苯基)丁酸甲酯。
[0108]
第五步:水解与酰氨化
[0109]
1.3g(3r)-n-叔丁氧羰基-3-氨基-4-(2,4,5-三氟苯基)丁酸甲酯,加入甲醇和质量百分数30%氢氧化锂水溶液,反应完全后,中和萃取,干燥浓缩后加1.0gdcc、1.2g3-(三氟甲基)-5,6,7,8-四氢-[1,2,4]三唑并[4,3-a]吡嗪盐酸盐和0.5g三乙胺,缓慢升至室温
后继续反应完毕后过滤除去生成的n,n’一二环己基脲,旋蒸除去溶剂,残留物用少量乙酸乙醋溶解后又有少旋蒸除去溶剂,残留物柱层析分离得白色泡沫状固体1.5g,收率为55.7%。
[0110]
通过对比可得,公开文件cn102153559a虽然合成路线短,但与本发明相比合成效率较低。
[0111]
本发明中所描述的具体实施例仅是对本发明精神作举例说明。本发明所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,但并不会偏离本发明的精神或者超越所附权利要求书所定义的范围。
[0112]
尽管对本发明已作出了详细的说明并引证了一些具体实施例,但是对本领域熟练技术人员来说,只要不离开本发明的精神和范围可作各种变化或修正是显然的。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。