1.本发明涉及药物化学制备领域,涉及一种重要中间体的制备方法,具体涉及4-氨基苯甲脒二盐酸盐的制备方法。
背景技术:
2.4-氨基苯甲脒二盐酸盐主要用作医药中间体,其是一种亲吸附剂的配基,与葡聚糖凝胶等结合在一起可以制成生物分离材料,会与一些特定的酶结合,可以模拟生物的抑制、抗体等作用;它是丝氨酸蛋白酶的竞争性抑制剂所以可以作为荧光探针来检测丝氨酸蛋白酶的活性部位。最近几年也有很多报道是将其作为原料药甲磺酸达比加群酯的重要中间体。
3.原研公司勃林格殷格翰在多次对甲磺酸达比加群酯进行工艺优化时发现使用4-氨基苯甲脒二盐酸盐可以大大简化合成路线节约成本并于专利wo2011061080 a1中报道了这一路线:
[0004][0005]
至今为止制备4-氨基苯甲脒二盐酸盐大致可以分成两条主流的合成路线:
[0006]
(一)将对氨基苯腈用羟胺做成肟,肟结构再用pd/c,pt/c,雷尼镍加氢还原肟基,如下所示:
[0007][0008]
该路线虽然有多种加氢金属催化剂可供选择,但事实上pd/c,pt/c都十分昂贵,在大规模放大中除非添加量很少,催化剂回收套用多次才具有成本优势,而雷尼镍价格低廉,却容易自燃增加不安全因素,另一方面加氢反应需要高压釜等特殊设备,无论是小试探索工艺条件还是放大都不利于操作。
[0009]
(二)pinner反应制备4-氨基苯甲脒盐酸盐,先在强酸或强碱中将氰基做成亚胺酯,再将亚胺酯用氨源氨解成脒基结构,原研公司勃林格殷格翰在专利us2014114089a1中报道了这一策略。路线如下:
[0010][0011]
该路线策略虽然简洁明了,理论上也可行,但是在实际上存在巨大致命缺陷,在50g克级小试中,反应3d,点板预估仅有约30%底物转化为亚胺酯,如此低的转化率显然在大生产上是不可行的。我们猜测是对位氨基的强供电子作用导致苯环上电子云密度过高,从而导致对氰基的加成反应难以进行。
技术实现要素:
[0012]
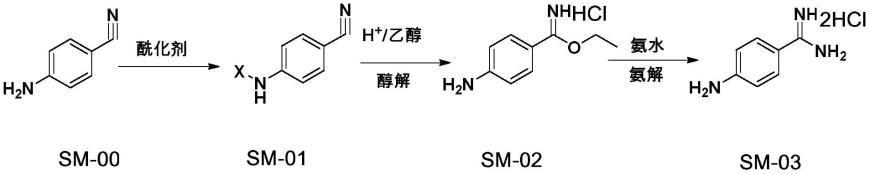
本技术提供了一种全新的4-氨基苯甲脒二盐酸盐的制备方法,该方法先通过酰化剂将对氨基苯甲腈氨基上的氢取代为吸电子基团,然后再通过醇解生成亚胺酯盐酸盐,可以有效提高对氨基苯甲腈到亚胺酯盐酸盐的转化率,最后进行氨解生成4-氨基苯甲脒。
[0013]
4-氨基苯甲脒二盐酸盐的制备方法,包括以下步骤:
[0014][0015]
其中,sm-00为对氨基苯甲腈,sm-02为亚胺酯盐酸盐,sm-03为4-氨基苯甲脒二盐酸盐;x为吸电基团,酰化剂为引入吸电子基团的试剂;
[0016]
(a)将对氨基苯甲腈溶解在溶剂中,加入酰化剂,在0~65℃搅拌反应2~4h,在40~70℃旋蒸移除溶剂,得到sm-01;
[0017]
(b)向sm-01中加入质量浓度为30%以上的氯化氢乙醇,在35~45℃下搅拌反应12~18h,在35~45℃旋干氯化氢乙醇溶液,得到亚胺酯盐酸盐;
[0018]
(c)向亚胺酯盐酸盐中加入反应溶剂,冰浴冷却至0~10℃滴加氨水,控制体系为碱性,在35~45℃反应2~4h,在75~85℃浓缩除去溶剂,得到4-氨基苯甲脒二盐酸盐粗品;
[0019]
(d)将4-氨基苯甲脒粗品和无水乙醇混合,搅拌状态下在10~20℃滴加浓盐酸,抽滤,将得到的固体在60~70℃真空干燥后得到4-氨基苯甲脒二盐酸盐精制品。
[0020]
本技术中的x为乙酰基、三氟乙酰乙基、丙酰基或二氯乙酰基;本技术中的酰化剂为乙酸酐、三氟乙酸酐、丙酸酐或二氯乙酰氯。
[0021]
具体地,当酰化剂为乙酸酐时,x为乙酰基;当酰化剂为三氟乙酸酐时,x为三氟乙酰基;当酰化剂为丙酸酐时,x为丙酰基;当酰化剂为二氯乙酰氯,x为二氯乙酰基。
[0022]
本技术步骤(a)中的溶剂是指dcm、三氯甲烷、1.2-二氯乙烷、氯苯或稀盐酸。
[0023]
优选地,本技术步骤(a)中的溶剂为dcm。
[0024]
本技术步骤(a)中,氯化氢乙醇选自大于30%(w/w)的氯化氢乙醇。30%浓度的氯化氢 乙醇是可以正常反应的最低浓度。低于这个浓度,酯水解副产物大幅增加。
[0025]
本技术步骤(c)中的碱性为ph=8。
[0026]
本技术步骤(c)中,所述反应溶剂为甲醇或氨水;所述氨水选自25%(w/w)的氨水。
[0027]
本技术步骤(c)中,加入浓盐酸后进行打浆处理:打浆、冷却、搅拌、抽滤和淋洗;重复上述打浆处理步骤0~1次,打浆处理的具体步骤为:加热到75~85℃打浆3h,冷却到15~
25℃,冷却速率:20~25℃/h,搅拌6~8h,抽滤。
[0028]
本技术步骤(a)中,加入酰化剂时加入三乙胺。
[0029]
本技术步骤(a)中,加入酰化剂时加入dmap。
[0030]
本技术带来的有益效果如下:
[0031]
1、本技术先通过酰化剂将对氨基苯甲腈氨基上的氢酰化为吸电子基团,然后再通过醇解生成亚胺酯盐酸盐,能将对氨基苯甲腈氨基到亚胺酯盐酸盐的转化率显著提高,使得整体的收率提高,从而利于大生产的进行。
[0032]
2、若酰化剂为酸酐,一般在低温下反应,因为酸酐与腈基可能发生加成。但是若酰化剂为丙酸酐,由于丙酸酐酰化能力弱,则需要提高温度才能使反应进行。
[0033]
3、三乙胺作为缚酸剂,使反应体系保持在一个合理的ph值有利于反应进行。dmap作为酰胺化反应或者酯化反应催化剂,视反应进行的困难程度加入适应的催化剂,以保证反应的顺利进行。
[0034]
4、本技术的(b)步骤中,30%浓度的氯化氢乙醇是可以正常反应的最低浓度。低于这个 浓度,酯水解副产物大幅增加;(c)步骤中,含有的残留盐酸量每批次是不等的,所以要确 认氨水一定过量,体系需要成碱性才可继续氨解反应,最优选地ph=8;精致步骤中,由于前 述sm-02亚胺酯盐酸盐带两当量的氯离子酸根,后续氨解会生成氯化铵。同时副反应也生成 氯化铵,因此需要打浆主要是除去氯化铵,必要时重复打浆步骤可以更好地除去氯化铵。
附图说明
[0035]
图1为4-氨基苯甲脒二盐酸盐碳谱。
[0036]
图2为4-氨基苯甲脒二盐酸盐氢谱。
[0037]
图3为反应副产物示意图。
具体实施方式
[0038]
为使本发明更加容易理解,下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而用于限制本发明的范围,下例实施例中未提及的具体实验方法,通常按常规实验方法进行。
[0039]
具体实施例中所用到的化学试剂都来自于阿达玛斯,百灵威,安耐吉,探索平台化学试剂公司,具体涉及到对氨基苯甲腈,乙酸酐,氯化氢乙醇,氨水,浓盐酸,三乙胺,二甲胺基吡啶,三氟乙酸酐,二氯乙酰氯,试剂纯度级别为分析级或者色谱级。
[0040]
反应/纯化所用到的溶剂都来自于大生产工业级溶剂库,具体涉及到二氯甲烷,甲醇,乙醇,领回后不用进一步旋蒸使用,直接下一步,若需要干燥除水则按照标准流程获取干燥溶剂。
[0041]
所用普通玻璃仪器来自于北京欣维尔玻璃仪器有限公司,具体使用涉及到三口烧瓶,茄形瓶,单口烧瓶,加料漏斗,恒压滴液漏斗。
[0042]
反应所用搅拌为磁力搅拌(探索平台)或者机械搅拌涉及到的具体为上海司乐仪器有限公司d2004w。控制反应温度通常用普通玻璃水银温度计(探索平台)或者数显温度计(日本tandd-tr-71wb)。
[0043]
光谱分析测试设备高效液相色谱仪型号:岛津-lc-2030c-plus;1h和
13
cnmr型号 brukeravanceiii hd 600mhz or am 400mhz(杭州师范大学-有机硅化学及材料技术教育部重点实验室)。
[0044]
实施例1
[0045]
(1)游离4-氨基苯甲脒的制备
[0046]
在一个500ml带机械搅拌的三口瓶中依次加入240g dcm,30g sm-00(对氨基苯甲腈), 冰浴冷至5
±
5℃,滴加28.5g乙酸酐,加毕,5
±
5℃在搅拌反应4h,tlc(pe:ea=1:1,uv:254)监控sm-00点消失后,60
±
5℃旋蒸移除dcm,得到一个灰白色固体sm-01,该固体不用进一步纯化,直接下一步。
[0047]
在一个500ml带机械搅拌的三口瓶中依次快速加入240g,~30%氯化氢乙醇,上述sm-01 灰白色固体,在40
±
5℃保证充分搅拌情况下反应12h,tlc(预先4m氢氧化钠水溶液解盐酸盐-dcm萃取-点有机相,pe:ea=1:1,uv:254以及茚三酮显色)监控sm-01点消失后,在 40
±
5℃旋干氯化氢乙醇得到淡黄色固体,该固体(亚胺酯盐酸盐-sm-02)不用进一步纯化,直接下一步。
[0048]
将装有上述固体500ml带机械搅拌的三口瓶中,预先加入60g甲醇分散,冰浴冷却到5
±
5℃在滴加120g氨水(~25%w/w),加毕用广谱ph试纸确认体系ph~8,升温40
±
5℃反应2h,tlc (pe:ea=1:1,uv:254)确定亚胺酯点消失视为反应终点,80
±
5℃浓缩除去溶剂后得到一个黄色固体粗品(游离对氨基苯甲脒)。
[0049]
(2)4-氨基苯甲脒二盐酸盐精制品制备
[0050]
在一个500ml带机械搅拌的三口瓶中依次加入上述sm-02游离态固体粗品,190g无水乙醇,60g水,搅拌状态下在15
±
5℃滴加48g浓盐酸,期间析出大量固体,开启加热80
±
5℃热打浆3h,关闭加热,冷却到20
±
5℃(冷却速率:20℃/h)搅拌6h,抽滤,滤饼用30g乙醇淋洗,得到白色固体。
[0051]
将上述第一次打浆后的白色固体置于一个500ml带机械搅拌的三口瓶中,加入45g水,开启加热80
±
5℃热打浆3h,关闭加热,冷却到20
±
5℃(冷却速率:20℃/h)在搅拌6h,,抽滤,滤饼用30g乙醇淋洗,得到的白色固体65
±
5℃真空干燥16h收料得到4-氨基苯甲脒盐酸盐成品34.5g,产品为白色或者类白色固体,总收率:65.2%,hplc纯度:99.89%。
[0052]1h nmr(400mhz,d2o)δ7.84(d,j=8.0hz,2h),7.50(d,j=8.0hz,2h);
13
c nmr(100 mhz,d2o)δ165.7,137.1,129.8,127.2,123.0;esi
:[m h]
=209.1。
[0053]
实施例2
[0054]
(1)游离4-氨基苯甲脒的制备
[0055]
在一个500ml带机械搅拌的三口瓶中依次加入210g dcm,15g sm3-00(对氨基苯甲腈), 19.2g三乙胺,1.56g dmap冰浴冷至5
±
5℃,滴加32.0g三氟乙酸酐,加毕,5
±
5℃在搅拌反应2h,tlc(pe:ea=1:1,uv:254)监控sm-00点消失后,45
±
5℃旋蒸移除dcm,得到一个棕黄色固体sm-01,该固体不用进一步纯化,直接下一步。
[0056]
在一个500ml带机械搅拌的三口瓶中依次快速加入90g,~30%氯化氢乙醇,上述sm-01 棕黄色固体,在40
±
5℃保证充分搅拌情况下反应12h,tlc(预先4m氢氧化钠水溶液解盐酸盐-dcm萃取-点有机相,pe:ea=1:5,uv:254以及茚三酮显色)监控sm-01点消失后,在 40
±
5℃旋干氯化氢乙醇得到淡黄色固体,该固体(亚胺酯盐酸盐-sm0-02)不用进一步
纯化,直接下一步。
[0057]
将装有上述固体500ml带机械搅拌的三口瓶中,预先加入30g甲醇分散,冰浴冷却到5
±
5℃在滴加105g氨水(~25%w/w),加毕用广谱ph试纸确认体系ph~8,升温40
±
5℃反应2h,tlc 确定亚胺酯点sm-02消失视为反应终点,80
±
5℃浓缩除去溶剂后得到一个黄色固体粗品(游离对氨基苯甲脒)。
[0058]
(2)4-氨基苯甲脒二盐酸盐精制品制备
[0059]
在一个500ml带机械搅拌的三口瓶中依次加入上述sm-03游离态固体粗品,94.5g无水乙醇,搅拌状态下在15
±
5℃滴加3.0g浓盐酸,期间析出大量固体,开启加热70
±
5℃热打浆3h,关闭加热,自然冷却到20
±
5℃,抽滤,滤饼用15g乙醇淋洗,得到淡黄色固体在65
±
5℃真空干燥12h收料得到4-氨基苯甲脒盐酸盐成品14.8g,产品为类白色或淡黄色固体,总收率:53.6%,hplc纯度:98.87%,核磁和质谱鉴定结果和实施例1一致。
[0060]
实施例3
[0061]
(1)游离4-氨基苯甲脒的制备
[0062]
在一个500ml带机械搅拌的三口瓶中依次加入270g 1.2mon/l稀盐酸,30g sm-00(对氨基苯甲腈),加热至60
±
5℃溶清,滴加132.2g丙酸酐,加毕,60
±
5℃在搅拌反应3h,期间析出大量白色固体,过滤得到sm-01备用。
[0063]
在一个500ml带机械搅拌的三口瓶中依次快速加入180g,~30%氯化氢乙醇,上述sm-01 白色固体,在40
±
5℃保证充分搅拌情况下反应12h,tlc(预先4m氢氧化钠水溶液解盐酸盐-dcm萃取-点有机相,pe:ea=1:1,uv:254以及茚三酮显色)监控sm-01点消失后,在40
±
5℃旋干氯化氢乙醇得到黄色固体,该固体(亚胺酯盐酸盐-sm-02)不用进一步纯化,直接下一步。
[0064]
将装有上述固体500ml带机械搅拌的三口瓶中加入120g氨水(~25%w/w),加毕用广谱ph试纸确认体系ph~8,升温至40
±
5℃反应2h,tlc(pe:ea=1:1,uv:254)确定亚胺酯点消失视为反应终点,80
±
5℃浓缩除去溶剂后得到一个黄色固体粗品(游离对氨基苯甲脒)。
[0065]
(2)4-氨基苯甲脒二盐酸盐精制品制备
[0066]
在一个500ml带机械搅拌的三口瓶中依次加入上述sm-02游离态固体粗品,189g无水乙醇,搅拌状态下滴加48g浓盐酸,期间析出大量固体,抽滤,得到的白色固体65
±
5℃真空干燥16h收料得到4-氨基苯甲脒盐酸盐成品21.7g,产品为白色或者类白色固体,总收率: 41.0%,hplc纯度:94.45%,核磁和质谱鉴定结果和实施例1一致。
[0067]
实施例4
[0068]
(1)游离4-氨基苯甲脒的制备
[0069]
在一个500ml带机械搅拌的三口瓶中依次180g dcm,30g sm-00(对氨基苯甲腈),38.5 g三乙胺,冰浴冷至5
±
5℃,滴加41.1g二氯乙酰氯,加毕在搅拌反应3h,加水萃取分液,水相再萃取(30ml),合并有机相,干燥,浓缩得到深棕色固体sm-01粗品,该粗品不用进一步纯化,直接下一步。
[0070]
在一个500ml带机械搅拌的三口瓶中依次快速加入180g~30%氯化氢乙醇,上述sm-01 深棕色固体,在40
±
5℃保证充分搅拌情况下反应18h,tlc(预先4m氢氧化钠水溶液解盐酸盐-dcm萃取-点有机相,pe:ea=1:1,uv:254以及茚三酮显色)监控sm-01点消失后,
在 40
±
5℃旋干氯化氢乙醇得到棕色固体,该固体(亚胺酯盐酸盐-sm-02)不用进一步纯化,直接下一步。
[0071]
将装有上述固体500ml带机械搅拌的三口瓶中加入120g氨水(~25%w/w),加毕用广谱ph试纸确认体系ph~8,升温40
±
5℃反应2h,tlc(pe:ea=1:1,uv:254)确定亚胺酯点消失视为反应终点,80
±
5℃浓缩除去溶剂后得到一个深棕色固体粗品(游离对氨基苯甲脒)。
[0072]
(2)4-氨基苯甲脒二盐酸盐精制品制备
[0073]
在一个500ml带机械搅拌的三口瓶中依次加入上述sm-02游离态固体粗品,120g无水乙醇,搅拌状态下滴加48g浓盐酸,期间析出大量固体,抽滤,得到的黄色固体65
±
5℃真空干燥12h收料得到4-氨基苯甲脒盐酸盐成品32.7g,产品为黄色固体,总收率:62.0%,hplc 纯度:95.4%,核磁和质谱鉴定结果和实施例1一致。
[0074]
实施例5
[0075]
在一个3000l带机械搅拌的搪玻璃反应釜中依次加入1840kg dcm,230kg sm-00(对氨基苯甲腈),冷乙二醇降温冷却至5
±
5℃在滴加218.5kg乙酸酐,加毕,5
±
5℃在搅拌反应 4h,tlc监控(硅胶板:gf254,展开剂:石油醚/乙酸乙酯=1/1,254nm紫外灯下检视原料对氨基苯甲腈斑点消失视为反应终点)sm-00点消失后,60
±
5℃旋蒸移除dcm,得到一个灰白色固体sm-01,该固体不用进一步纯化,直接下一步。
[0076]
在上述装有酰化产物固体的3000l搪玻璃反应釜中加入1840kg,~30%氯化氢乙醇,先 5
±
5℃在搅拌2h,再升温40
±
5℃保证充分搅拌情况下反应12h,tlc监控(硅胶板:gf254,展开剂:石油醚/乙酸乙酯=1/1,254nm紫外灯下检视sm-01乙酰化产物斑点消失视为反应终点)sm-01点消失后,保证40
±
5℃旋干氯化氢乙醇,得到黄色固体亚胺酯,该固体(亚胺酯盐酸盐-sm-02)不用进一步纯化,直接下一步。
[0077]
将装有上述固体3000l带机械搅拌的搪玻璃反应釜中,预先加入460kg甲醇分散,冷乙二醇夹套冷却到5
±
5℃在滴加920kg 25%氨水溶液,加完升温40
±
5℃反应2h,tlc(硅胶板: gf254,展开剂:石油醚/乙酸乙酯=1/1,254nm紫外灯下检视sm-02亚胺酯产物斑点消失视为反应终点,产物点在底线上)确定亚胺酯点消失后,85
±
5℃,最终真空度≤-0.090mpa除去溶剂甲醇和水得到一个黄色固体粗品(游离对氨基苯甲脒)。
[0078]
在装有上述固体3000l带机械搅拌的搪玻璃反应釜中,预先加入460kg水升温80
±
5℃分散搅拌2h,加入1449kg无水乙醇、冷乙二醇,夹套冷却至10
±
5℃,搅拌状态下在10
±
5℃加入368kg浓盐酸,期间析出大量固体将其加热80
±
5℃搅拌3h,降温速率:20℃/h,降4 小时至20
±
5℃析晶6h,离心得到白色固体湿品605kg。
[0079]
将上述第一次打浆后的白色固体置于一个3000l带机械搅拌的搪玻璃反应釜中345kg水,开启加热80
±
5℃热打浆3h,降温速率:20℃/h,降4小时至20
±
5℃,析晶6h离心,离心固体用230kg乙醇淋洗,得到白色固体湿品276kg,得到的白色固体65
±
5℃真空干燥16h收料得到sm003二盐酸盐成品257.5kg,产品为白色固体,总收率:63.4%,hplc纯度:99.67%。
[0080]
对比例1
[0081]
在一个带有机械搅拌的1000ml三口瓶中依次加入sm-00(对氨基苯甲腈)50g,400g,~30%氯化氢乙醇,先5
±
5℃在搅拌2h,再升温45
±
5℃保证充分搅拌情况下反应3d,
tlc监控(硅胶板:gf254,展开剂:石油醚/乙酸乙酯=1/1,254nm紫外灯下检视sm-00)反应3d,sm3-00 仍旧大量残留,保证45
±
5℃旋干氯化氢乙醇,得到黄色固体亚胺酯混合物,该固体(亚胺酯盐酸盐sm-02)不用进一步纯化,直接下一步。
[0082]
向装有上述固体的三口瓶中,预先加入100g甲醇分散,冷却到5
±
5℃在滴加200g 25%氨水溶液,加完升温40
±
5℃反应2h,tlc(硅胶板:gf254,展开剂:石油醚/乙酸乙酯=1/1, 254nm紫外灯下检视sm-02亚胺酯产物斑点消失视为反应终点,产物点在底线上)确定亚胺酯点消失后,85
±
5℃,最终真空度≤-0.080mpa除去溶剂甲醇和水得到一个黄色固体粗品(游离对氨基苯甲脒含大量未反应完全的初始物料sm-00)。
[0083]
向装有上述固体的1000ml三口瓶中,预先加入100g水升温80
±
5℃分散搅拌2h,加入315g无水乙醇,冷却至10
±
5℃,搅拌状态下在10
±
5℃加入80g浓盐酸,期间析出大量固体将其加热80
±
5℃搅拌3hrs,降温速率:20℃/h,降4小时至20
±
5℃析晶6h,抽滤, 抽滤固体用50g无水乙醇淋洗,得到淡黄色固体湿品47g。
[0084]
将上述离心后的固体置于一个1000ml带机械搅拌的三口瓶中,加入100g水,315g无水乙醇,开启加热80
±
5℃热打浆3h,降温速率:20℃/h,降4小时至20
±
5℃,析晶6h抽滤,抽滤固体50g乙醇淋洗,得到白色固体对氨基苯甲脒二盐酸盐粗品。将上述固体倒入到 250ml三口瓶中,加入25g水,80
±
5℃搅拌3h,降温速率:20℃/h,降4小时至20
±
5℃析晶6h,抽滤,抽滤固体用50g无水乙醇淋洗得到的白色固体产品于65
±
5℃真空干燥16h收料得到4-氨基苯甲脒二盐酸盐成品17g,产品为白色固体,总收率:19.3%,hplc纯度:96.25%。
[0085]
实施例1~5和对比例1的对比如下表所示:
[0086]
实施例对氨基苯甲脒二盐酸盐总收率实施例165.2%实施例253.6%实施例341.0%实施例462.0%实施例563.4%对比例119.3%
[0087]
试验例1:氯化氢乙醇对比试验
[0088]
以下试验例仅氯化氢乙醇浓度(质量浓度)变化,其他条件同实施例1:
[0089][0090][0091]
由上述试验可知,氯化氢乙醇的浓度太低,酯水解副产物会增多。当氯化氢乙醇的浓度大于30%时,酯水解副产物能够控制在一个较低的水平。
[0092]
以上所述的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范
围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案作出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。