1.本发明属于药物化合物技术领域,具体涉及一种肉桂酸酯衍生物结构、其制备方法及其抗肿瘤活性与制药应用。
背景技术:
2.天然有机物酚酸是植物降解的产物,在植物中发现的两大类酚酸包括苯甲酸和肉桂酸(3-苯基丙烯酸)及其衍生物。最常见的肉桂羟基衍生物是对香豆酸(4-羟基肉桂酸)和咖啡酸(3,4-二羟基肉桂酸)。肉桂酸是莽草酸途径的关键中间产物,是类黄酮或木质素合成的前体。肉桂酸衍生物是植物的次生代谢产物,在植物的生长、发育、繁殖和抗病等方面发挥着重要作用。肉桂酸衍生物由于在植物中广泛存在,毒性低,生物活性高,被认为是食品添加剂或药理活性化合物。苯环上取代的丙烯酸基团使肉桂酸具有顺式(z)或反式(e)构型,其中反式(e)构型最为常见,肉桂酸可通过苯丙氨酸的酶促脱氨反应制备。肉桂酸的结构如下:
3.肉桂酸衍生物在治疗癌症、细菌感染、糖尿病和神经系统疾病等方面的作用已被报道。除了天然存在于植物中的肉桂酸衍生物外,苯环和丙烯酸基团的存在使其修饰合成肉桂酸衍生物成为可能。肉桂酸衍生物不仅是二苯乙烯、苯乙烯等化合物的中间体,还具有抗肿瘤、抗菌、抗真菌、抗炎、神经保护和抗糖尿病等活性。已有文献报道的肉桂酸衍生物有阿魏酸咖啡酸绿原酸以及deng等人从植物内生真菌pyronema sp中提取的等。这些肉桂酸衍生物的结构修饰涉及苯环、丙烯基和羧基连接子等部分。据报道,与用于治疗慢性或传染病的标准药物相比,其中一些衍生物在体外更有效,因此使它们成为非常有前途的治疗药物。
4.肉桂酸衍生物的生物活性改变归因于取代基的性质和位置,例如苯环上取代基的性质和位置影响衍生物的抗菌、抗癌和抗氧化活性,羧基连接子的长度也影响衍生物的生物活性。常规治疗药物的耐药性和缺乏具有低副作用的治疗方法来控制肿瘤、微生物生长、神经系统疾病等现实因素推动了基于肉桂酸的治疗性化合物的开发研究。
技术实现要素:
5.本发明提供了一种肉桂酸酯衍生物,其具有有价值的药理性质,特别是抑制肿瘤生长、加速细胞凋亡和抑制细胞活力。
6.本发明还进一步提供了所述肉桂酸酯衍生物的制备方法。
7.具有式i结构的化合物,或者其立体异构体、药物可接受的盐或多晶型:
[0008][0009]
其中:
[0010]
r1为苯环上的氢或取代基,r1为取代基时可相同或不同,独立的选自单取代或多取代,r1独立的选自氢、卤素、1-4个碳原子的烷基、1-4个碳原子的烷氧基、1-4个碳原子的卤代烷烃、1-4个碳原子的烷苯基、0-4个碳原子的烷氧苯基、未取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物或被叔丁氧羰基等取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物中的一个或更多个基团;
[0011]
r2和r3相同或不同,独立的选自氢、未取代或取代的苯基、萘基、蒽基、菲基,其中的取代基被独立地选自卤素、1-4个碳原子的烷基、1-4个碳原子的烷氧基、1-4个碳原子的卤代烷烃、1-4个碳原子的烷苯基、0-4个碳原子的烷氧苯基、未取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物或被叔丁氧羰基等取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物中的一个或更多个基团;
[0012]
r4独立的选自氢、甲基、二取代甲基、三取代甲基、未取代或取代的1-4个碳原子的烷苯基、萘基、蒽基、菲基,其中1-4个碳原子的烷苯基、萘基、蒽基、菲基的取代基被独立地选自卤素、1-4个碳原子的烷基、1-4个碳原子的烷氧基、1-4个碳原子的卤代烷烃、1-4个碳原子的烷苯基、0-4个碳原子的烷氧苯基或卤素等取代的0-4个碳原子的烷氧苯基、未取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物或被叔丁氧羰基等取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物中的一个或更多个基团;
[0013]
表示单键或双键;
[0014]
优选,r1为苯环上的氢或取代基,r1为取代基时选自单取代或二取代,r1选自氢、1-4个碳原子的烷氧基、1-4个碳原子的烷氧苯基、未取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物或被叔丁氧羰基等取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物中的一个或更多个基团;
[0015]
优选,r2和r3相同,均选自氢;
[0016]
优选,表示单键或双键;
[0017]
优选,r4选自三取代甲基、未取代的1-4个碳原子的烷苯基或取代的1-4个碳原子的烷苯基,其中取代基被独立地选自1-4个碳原子的烷基、1-4个碳原子的烷氧基、0-4个碳原子的烷氧苯基或卤素等取代的0-4个碳原子的烷氧苯基、未取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物或被叔丁氧羰基等取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物中的一个或更多个基团。
[0018]
在本发明的一个实施方案中,其中r1为苯环上的氢或取代基,r1为取代基时选自单
取代或二取代,r1选自氢、甲氧基、未取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物或被叔丁氧羰基等取代的包含氧氮硫的饱和或不饱和五元六元杂环化合物中的一个或更多个基团,r2和r3均选自氢,选自单键或双键,r4选自三取代甲基、未取代的1-4个碳原子的烷苯基或被独立地选自烷氧基、烷氧苯基或卤素等取代的烷氧苯基、哌啶基、吡啶基、叔丁氧羰基取代哌啶基或叔丁氧羰基取代吡啶基中的一个或更多个基团取代的1-4个碳原子的烷苯基,其具有式ii结构:
[0019]
根据本发明的一个实施方案,优选化合物为式ii所示的化合物,其中r1独立的选自氢、对位单取代间对位二取代邻位单取代r4独立的选自独立的选自独立的选自示例性化合物包括表1中阐述的化合物及其药物可接受的盐或其溶剂合物:
[0020]
表1
[0021]
[0022][0023]
本发明还提供式i、ii和iii化合物的制备方法:式i、ii和iii中的部分化合物由左侧酸部分ra进一步反应生成酰氯,然后与右侧醇部分rb缩合而成,
[0024][0025]
根据本发明的示例性化合物中左侧酸部分ra包括表2阐述的中间体化合物:
[0026]
表2
[0027][0028]
中间体化合物ra1和ra2可以按照本领域已知的合成方法制备,例如:(ra1)in quest of small-molecules as potent non-competitive inhibitors against influenza,bioorg.chem.,2021,114,105139;(ra2)n-hydroxyphthalimide catalyzed aerobic oxidation of aldehydes under continuous flow conditions,advanced synthesis&catalysis,2022,364,1998-2008。
[0029]
根据本发明的示例性化合物中右侧醇或胺部分包括表3阐述的中间体化合物:
[0030]
表3
[0031]
[0032][0033]
其中,中间体化合物rb1~rb8可按照本领域已知的合成方法制备,例如:(rb4)cn 109516914a,2019.03.26;(rb7)potent human glutaminyl cyclase inhibitors as potential anti-alzheimer’s agents:structure-activity relationship study of arg-mimetic region,bioorgan.med.chem.,2018,26,1035-1049。
[0034]
因此本发明还提供上述中间体化合物rb9和rb10的制备方法,其中rb9制备方法如下:rb10的制备方法如下:
[0035][0036]
在本发明的一个实施方案中,化合物12和13的制备方法如下:
[0037]
[0038]
在本发明的一个实施方案中,化合物14、15和16的制备方法如下:
[0039][0040]
除特别说明之外,上述制备方法中已有文献报道的化合物均可以采用本领域已知的反应方法和条件进行。
[0041]
本发明还提供一种药物组合物,其包含本发明的式i化合物或其溶剂化物作为活性成分,任选地还含有一种或多种药学上可接受的载体。所述药学上可接受的载体是制药领域中常用或已知的各种辅料,包括但不限于:稀释剂、粘合剂、抗氧化剂、ph调节剂、防腐剂、润滑剂、崩解剂等。
[0042]
在本发明的一种实施方式中,所述药物组合物用于治疗或预防癌症,抑制肿瘤生长、加速细胞凋亡和抑制细胞活力。
[0043]
根据本发明,所述癌症包括原发性和继发性癌症,包括但不限于肝癌、肺癌、淋巴癌和甲状腺癌等,优选,所述癌症为结肠癌。
[0044]
所述药物组合物中含有式i化合物的量(以式i化合物计)为0.1-1000mg,优选1-500mg,更优选为5-100mg。
[0045]
所述药物组合物中式i化合物(以式i化合物计)占药物组合物的质量百分比为0.01%-95%,根据剂型不同例如可以为0.1%-10%,0.3~5%,或者10%-90%,优选为20%-80%,更优选为30%-70%等含量范围。
[0046]
所述药物组合物的剂型可以是口服剂的形式,例如片剂、胶囊、丸剂、粉剂、颗粒剂、悬浮剂、糖浆剂等;也可以是注射给药的剂型,例如注射液、粉针剂等,通过静脉内、腹膜内、皮下或肌肉内的途径注射给药。所有使用的剂型形式都是药学领域普通技术人员所熟知的。例如所述药物组合物可以为注射液,式i化合物在注射液中的浓度可以为1-15mg/ml,例如5mg/ml、10mg/ml、12.5mg/ml等。
[0047]
所述药物组合物的施用途径包括但不限于:口服的;含服的;舌下的;透皮的;肺的;直肠的;肠胃外的,例如,通过注射,包括皮下的、真皮内的、肌内的、静脉内的;通过植入储库或储液器。
[0048]
式i化合物的施用剂量(以式a化合物计)将取决于接受者的年龄、健康和体重,联用药物的种类,治疗频率,给药途径等。药物可以单一日剂量施用,每天给药一次、每两天给药一次、每三天给药一次、每四天给药一次,或者总日剂量以每天两次、三次或四次的分开剂量施用。式i化合物用药量(以式i化合物计)为0.01-100mg/kg/天,优选为0.1-10mg/kg/天,例如为0.5mg/kg/天,1mg/kg/天、2mg/kg/天、5mg/kg/天等等。
[0049]
所述药物组合物可以和其他的治疗剂联合应用给药或者制成组合药物。所述其他治疗剂根据疾病和病症类型不同,可以是其他的治疗癌症的药物等。
[0050]
其他的治疗癌症的药物包括但不限于:紫杉类(多西他赛、紫杉醇)、铂类(奥沙利铂、顺铂)、氟尿嘧啶类(希罗达、替吉奥)、蒽环类(多柔吡星、吡柔吡星)、吉菲替尼、索拉菲尼、抗生素、血容量扩充剂、血管活性药物、糖皮质激素、血制品、血糖控制药物、抗凝药等。
[0051]
本发明提供式i化合物在制备治疗或预防癌症的药物中的应用。所述药物能够抑制肿瘤生长、加速细胞凋亡和抑制细胞活力。
[0052]
本发明提供式i化合物和其他的治疗癌症的药物联合在制备治疗或预防癌症的药物中的用途。
[0053]
本发明提供式i化合物和其他的治疗癌症的药物联合在制备抑制癌症患者的肿瘤生长的药物中的用途。
[0054]
本发明提供式i化合物和其他的治疗癌症的药物联合在制备加速癌症患者的肿瘤细胞凋亡的药物中的用途。
[0055]
本发明提供式i化合物和其他的治疗癌症的药物联合在制备抑制癌症患者的肿瘤细胞活力的药物中的用途。
[0056]
本发明提供式i化合物在制备和其他的治疗癌症的药物联合治疗或预防癌症药物中的用途。
[0057]
本发明提供一种治疗或预防癌症,抑制肿瘤生长、加速细胞凋亡和抑制细胞活力的方法,其特征在于,对有需要的患者施用治疗有效量的式i化合物或其溶剂化物、或者含有式i化合物或其溶剂化物的药物组合物。
[0058]
本发明式i化合物具有明确且高效的癌症治疗和预防效果,能够抑制肿瘤生长、加速细胞凋亡和抑制细胞活力,不具有细胞毒性。因此在治疗用途的同时保证用药的安全性。式i化合物结构明确,利于制备和质量控制。
附图说明
[0059]
图1cck-8检测hct-8细胞活力实验结果
[0060]
图2cck-8检测hct116细胞活力实验结果
具体实施方式
[0061]
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0062]
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0063]
pcc:氯铬酸吡啶盐;(dppf)2pdcl2:[1,1'-双(二苯基膦基)二茂铁]二氯化钯;pd(oac)2:醋酸钯;pd/c:钯碳;pph3:三苯基磷;tbab:四丁基溴化铵;nabh4:硼氢化钠;bppo:n1,n2双([1,1'-联苯]-2-基)乙二酰胺;tmsbr:三甲基溴硅烷;(cocl)2:草酰氯;tfa:三氟乙酸;dmf:n,n-二甲基甲酰胺;pd(pph3)4:四三苯基膦钯。
[0064]
实施例1 ra系列化合物的制备
[0065]
1、肉桂酸(ra1)的制备方法
[0066][0067]
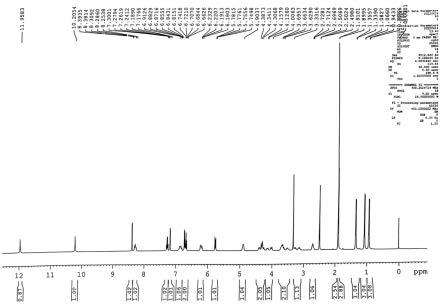
在室温下,50ml dmf中依次加入苯甲醛(5.00ml,49.06mmol)、丙二酸(15.32g,147.31mmol)以及吡啶(4.00ml,49.72mmol)。95℃搅拌过夜后tlc监测,待反应完全后用饱和nahco3溶液/二氯甲烷体系萃取。收集有机层后加入过量无水硫酸钠干燥,随后过滤并减压浓缩,经硅胶柱色谱法纯化(石油醚:乙酸乙酯=2:1,体积比),得到白色固体肉桂酸(ra1)(6.71g,92%)。
[0068]1h nmr(400mhz,cdcl3)δ12.07(bs,1h),7.80(d,j=16.0hz,1h),7.58
–
7.51(m,2h),7.42
–
7.36(m,3h),6.46(d,j=16.0hz,1h).
13
c nmr(100mhz,cdcl3)δ172.86,147.25,134.16,130.88,129.09(2c),128.51(2c),117.48.
[0069]
实施例2 rb系列化合物的制备
[0070]
1、(3-(苯氧基甲基)苯基)甲醇(rb2)的制备方法
[0071][0072]
将间溴甲基苯甲酸甲酯(2.01g,8.77mmol),苯酚(0.98g,10.41mmol),tbab(1.40g,4.35mmol)以及磷酸钾(2.80g,13.19mmol)依次加入到100ml圆底烧瓶中,加入纯水(10ml)作溶剂,室温搅拌24h。tlc监测反应结束后,0℃下加入饱和nahco3溶液碱化反应液,用dcm与饱和nacl溶液萃取,收集dcm层,加入无水na2so4干燥,过滤,收集滤液并浓缩。通过柱层析色谱法进行分离纯化(石油醚:乙酸乙酯=30:1),减压浓缩,得到白色粉末状化合物rb2-1(2.08g,98%)。
[0073]1h nmr(400mhz,cdcl3)δ8.10(s,1h),7.98(d,j=7.7hz,1h),7.60(d,j=7.6hz,1h),7.43(s,1h),7.27(t,j=7.8hz,2h),6.95
–
6.93(m,3h),5.05(s,2h),3.89(s,3h).
13
c nmr(100mhz,cdcl3)δ166.88,158.61,137.65,131.89,130.56,129.59,129.16,128.73,128.55,121.21,114.91,69.35,52.18.
[0074]
将化合物rb2-1(0.65g,2.70mmol),氯化锂(0.11g,2.60mmol)加入到100ml圆底烧瓶中,加入thf(10ml)将其溶解,0℃下缓慢加入硼氢化钠(0.61g,17.02mmol)的thf溶液,65℃下加热回流15min,加入甲醇(8ml),65℃下反应3h。tlc监测反应结束后,冷却至室温,加入饱和nh4cl溶液(10ml)淬灭反应,用dcm与饱和nacl溶液进行萃取,收集dcm层,加入无水na2so4干燥,过滤,收集滤液并浓缩。通过柱层析色谱法分离纯化(石油醚:乙酸乙酯=3:1),减压浓缩,得到白色粉末状化合物(3-(苯氧基甲基)苯基)甲醇(rb2)(0.54g,94%)。
[0075]1h nmr(400mhz,cdcl3)δ7.38(s,1h),7.33(s,2h),7.28
–
7.26(m,3h),6.95(t,j=
7.2hz,3h),5.03(s,2h),4.62(s,2h),3.78(s,1h).
13
c nmr(100mhz,cdcl3)δ158.77,141.36,137.43,129.56,128.84,126.77,126.58,126.04,121.07,114.90,69.85,65.08.
[0076]
2、(3-(3-氟苯氧基)苯基)甲醇(rb4)的制备方法
[0077]
rb3-rb6为同一系列的结构类似物,可按照相似方法合成获得,以rb4为例详述其制备方法:
[0078][0079]
依次取3-羟基苄醇(1.00g,8.06mmol)、间溴氟苯(1.35ml,12.09mmol)、bppo(0.03g,0.08mmol)、k3po4(3.42g,16.13mmol)以及cui(0.02g,0.10mmol)于100ml三颈烧瓶中并加入20ml dmf溶解,ar2保护,90℃搅拌过夜,tlc监测,反应停止后应用乙酸乙酯/饱和食盐水体系萃取,收集有机层后加入过量无水硫酸钠干燥0.5h随后过滤并减压浓缩,经硅胶柱色谱法纯化(石油醚:乙酸乙酯=20:1),得到无色液体状(3-(3-氟苯氧基)苯基)甲醇(rb4)(0.50g,28%),。
[0080]1h nmr(400mhz,cdcl3)δ7.30(t,j=7.9hz,1h),7.23(td,j=8.3,6.7hz,1h),7.09(d,j=7.6hz,1h),7.00(t,j=2.0hz,1h),6.92(dd,j=8.1,2.5hz,1h),6.79-6.74(m,2h),6.67(dt,j=10.2,2.4hz,1h),4.60(s,2h),2.66(bs,1h).
13
c nmr(100mhz,cdcl3)δ158.84,158.73,156.62,143.23,130.63,130.53,130.08,122.39,118.53,117.81,114.15,114.12,64.65.
[0081]
3、rb7和rb8的制备方法
[0082][0083]
1)rb7的制备
[0084]
依次取3-溴苯甲醇(1.40ml,11.68mmol)、n-boc-1,2,5,6-四氢吡啶-4-硼酸频哪醇酯(3.00g,9.71mmol)、(dppf)2pdcl2(0.07g,96.99μmol)以及k3po4(6.17g,29.10mmol)于50ml三颈烧瓶中并加入20ml 二氧六环与水的混合溶液(二氧六环:水=4:1)溶解,ar2保护,80℃搅拌4h,tlc监测,反应完毕后应用二氯甲烷/饱和食盐水体系萃取,收集有机层后加入过量无水硫酸钠干燥0.5h随后过滤并减压浓缩,经硅胶柱色谱法纯化(石油醚:乙酸乙酯=10:1,体积比),得到无色液体状3-(4-(1-叔丁氧羰基-2,3,6-三氢吡啶基))苄醇(rb7)(2.30g,82%)。
[0085]1h nmr(400mhz,cdcl3)δ7.38
–
7.29(m,2h),7.16(t,j=7.9hz,1h),7.11(d,j=7.9hz,1h),4.23(s,2h),2.77(t,j=12.8hz,2h),2.60(td,j=12.2,3.3hz,1h),1.85
–
1.71(m,2h),1.65
–
1.51(m,2h),1.51
–
1.40(m,9h).
13
c nmr(100mhz,cdcl3)δ154.86,148.19,130.20,130.08,129.53,125.56,122.68,79.62,44.42,42.56,33.10,28.58(3c).
[0086]
2)rb8的制备
[0087][0088]
取化合物rb7(0.50g,1.73mmol)以及10%的pd/c(0.05g)于50ml三颈烧瓶中,加入20ml乙酸乙酯溶解,经重复换气后接入氢气球,室温搅拌过夜,tlc监测,反应完全后垫硅藻土过滤,减压浓缩后上柱,经硅胶柱色谱法纯化后得到3-(4-(1-叔丁氧羰基)哌啶基)苄醇(rb8)(0.49g,97%)。
[0089]1h nmr(400mhz,cdcl3)δ7.19(t,j=7.8hz,1h),7.06
–
6.91(m,3h),4.24(s,2h),2.78(t,j=13.5hz,2h),2.60(tt,j=12.1,3.6hz,1h),2.33(s,3h),1.82(d,j=3.6hz,1h),1.79(d,j=3.3hz,1h),1.61(qd,j=12.7,4.3hz,2h),1.48(s,9h).
13
c nmr(100mhz,cdcl3)δ154.86,148.19,130.20,130.08,129.53,125.56,122.68,79.62,44.13,42.56(2c),33.10,28.58(3c).
[0090]
4、(3-甲氧基-5-(苯氧基甲基)苯基)甲醇(rb9)的制备方法
[0091][0092]
1)rb9-1的制备
[0093][0094]
将5-甲氧基-异邻苯二甲酸甲酯(0.63g,2.81mmol)放置于100ml圆底烧瓶中,加入甲醇(10ml)使其溶解,0℃下滴加硼氢化钠(1.12g,29.60mmol)的甲醇溶液,室温下反应4h。tlc监测反应结束后,在0℃下,加入饱和nh4cl溶液淬灭硼氢化钠,用dcm与饱和nacl溶液进行萃取,收集dcm层,加入无水na2so4干燥,过滤,收集滤液并浓缩。通过柱层析色谱法分离纯化(石油醚:乙酸乙酯=5:1),通过旋转蒸发仪进行减压浓缩,得到白色粉末状化合物rb9-1(0.39g,71%)。
[0095]1h nmr(400mhz,cdcl3)δ7.55(s,1h),7.41(s,1h),7.08(s,1h),4.65(s,2h),3.88(s,3h),3.81(s,3h),3.10(s,1h).
13
c nmr(100mhz,cdcl3)δ167.06,159.61,143.00,131.16,120.09,117.43,113.04,64.14,55.36,52.18.
[0096]
2)rb9-2的制备
[0097][0098]
将化合物rb9-1(0.4ml,2.37mmol)加入到5ml茄形瓶中,加入三甲基溴硅烷(0.7ml,5.01mmol),室温下反应12h。tlc监测反应结束后,直接通过柱层析色谱法进行分离纯化(石油醚:乙酸乙酯=50:1),减压浓缩,得到淡黄色油状化合物rb9-2(0.61g,99%)。
[0099]1h nmr(400mhz,cdcl3)δ7.66(s,1h),7.49(s,1h),7.12(s,1h),4.47(s,2h),3.92(s,3h),3.85(s,3h).
13
c nmr(100mhz,cdcl3)δ166.39,159.84,139.42,131.96,122.47,119.78,114.27,55.61,52.32,32.47.
[0100]
3)rb9-3的制备
[0101][0102]
将化合物rb9-2(0.62g,2.39mmol),苯酚(0.26g,2.76mmol),tbab(0.37g,1.15mmol)以及磷酸钾(0.73g,3.42mmol)按顺序加入到100ml圆底烧瓶中,加入纯水(15ml)作溶剂,室温反应24h。tlc监测反应结束后,0℃下加入饱和na2co3溶液碱化反应液,用dcm与饱和nacl溶液进行萃取,收集dcm层,加入无水na2so4干燥,过滤,收集滤液并浓缩。通过柱层析色谱法分离纯化(石油醚:乙酸乙酯=20:1),通过旋转蒸发仪进行减压浓缩,得到白色化合物rb9-3(0.52g,80%)。
[0103]1h nmr(400mhz,cdcl3)δ7.70(s,1h),7.51(s,1h),7.28(t,j=7.9hz,2h),7.19(s,1h),6.97
–
6.95(m,3h),5.05(s,2h),3.91(s,3h),3.84(s,3h).
13
c nmr(100mhz,cdcl3)δ166.88,159.93,158.56,139.12,131.71,129.56,121.21,120.82,120.52,118.27,115.36,114.92,113.66,69.29,55.56,52.28.
[0104]
4)rb9的制备
[0105][0106]
将化合物rb9-3(0.50g,1.84mmol),氯化锂(0.08g,1.89mmol)加入到100ml圆底烧瓶中,用thf(5ml)使其溶解,0℃下缓慢加入硼氢化钠(0.72g,19.0mmol)的thf溶液,70℃下加热回流15min,加入甲醇(3ml),继续在70℃下下反应12h。tlc监测反应结束后,室温冷却,加入饱和nh4cl溶液(10ml)淬灭反应,用dcm与饱和nacl溶液进行萃取,收集dcm层,加入无水na2so4干燥,过滤,收集滤液并浓缩。通过柱层析色谱法进行分离纯化(石油醚:乙酸乙酯=30:1),减压浓缩,得到白色粉末状化合物rb9(0.30g,67%)。
[0107]1h nmr(400mhz,cdcl3)δ7.26(t,j=7.7hz,2h),6.99
–
6.89(m,4h),6.87(s,1h),
6.83(s,1h),4.97(s,2h),4.59(s,2h),3.76(s,3h),2.53(s,1h).
13
c nmr(100mhz,cdcl3)δ160.12,158.75,142.99,138.93,129.54,121.08,118.03,114.92,112.22,111.80,69.77,64.95,55.34.
[0108]
5、(3-(2,6-二氟苯氧基)苯基)甲醇(rb10)的制备方法
[0109][0110]
依次取3-羟基苄醇(1.00g,8.06mmol)、1-溴-2,6-二氟苯(1.36ml,12.05mmol)、bppo(0.03g,0.08mmol)、k3po4(3.42g,16.13mmol)以及cui(0.02g,0.11mmol)于100ml三颈烧瓶中并加入20ml dmf溶解,ar2保护,90℃搅拌过夜,tlc监测,反应停止后应用乙酸乙酯/饱和食盐水体系萃取,收集有机层后加入过量无水硫酸钠干燥0.5h随后过滤并减压浓缩,经硅胶柱色谱法纯化(石油醚:乙酸乙酯=20:1,体积比),得到(3-(2,6-二氟苯氧基)苯基)甲醇(rb10)(0.27g,14%),无色液体。
[0111]1h nmr(400mhz,cdcl3)δ7.27(t,j=7.9hz,1h),7.14(td,j=8.3,6.2hz,1h),7.07(d,j=7.6hz,1h),6.95(s,1h),6.90
–
6.77(m,2h),6.65(dt,j=8.3,1.3hz,1h),4.62(s,2h),1.65(bs.,1h).
19
f nmr(376mhz,cdcl3)δ-104.51.
13
c nmr(100mhz,cdcl3)δ156.84,143.35,130.19,128.77,128.68,122.37,117.78,116.95,115.42,115.39,111.60,111.37,64.95.
[0112]
实施例3化合物1-15的制备方法
[0113]
1、化合物1和2的制备
[0114]
1)(e)-(3-苯氧基甲基)肉桂酸苄酯(化合物1)的制备
[0115][0116]
取化合物ra1(0.50g,3.38mmol)于烘干的50ml圆底烧瓶中,加入20ml无水dcm溶解,滴入1滴干燥的dmf做催化剂,冰浴并缓慢加入草酰氯(0.25ml,2.95mmol)搅拌反应40min,tlc监测,待反应完全后加入干燥的三乙胺(0.70ml,5.05mmol)以及rb2(0.52ml,2.76mmol),室温反应1h,反应完全后加水(5ml)淬灭,二氯甲烷/饱和食盐水体系萃取,收集有机层后加入过量无水硫酸钠干燥0.5h随后过滤并减压浓缩,经硅胶柱色谱法纯化,得到白色固体(e)-(3-苯氧基甲基)肉桂酸苄酯(化合物1)(0.89g,两步产率94%)。
[0117]1h nmr(400mhz,cdcl3)δ7.73(d,j=16.0hz,1h),7.54
–
7.46(m,3h),7.41
–
7.38(m,2h),7.38
–
7.35(m,4h),7.28(t,j=8.0hz,2h),7.01
–
6.92(m,3h),6.48(d,j=16.0hz,1h),5.26(s,2h),5.07(s,2h).
13
c nmr(100mhz,cdcl3)δ166.77,158.77,145.30,137.62,136.54,134.41,130.41,130.29,129.55(2c),128.94(2c),128.17(2c),127.84(2c),127.37,127.28,121.09,117.88,114.91,69.73,66.19.
[0118]
2)(e)-3-甲氧基-(5-苯氧基甲基)肉桂酸苄酯(化合物2)的制备
[0119][0120]
合成步骤同化合物1,即ra1和草酰氯反应生成的酰氯化合物再与rb9反应最终获得(e)-3-甲氧基-(5-苯氧基甲基)肉桂酸苄酯(化合物2)(0.14g,两步产率93%)。
[0121]1h nmr(400mhz,cdcl3)δ7.73(d,j=16.0hz,1h),7.56
–
7.47(m,2h),7.42
–
7.33(m,3h),7.32
–
7.24(m,2h),7.06(s,1h),7.01
–
6.92(m,4h),6.90(s,1h),6.49(d,j=16.0hz,1h),5.23(s,2h),5.04(s,2h),3.82(s,3h).
13
c nmr(100mhz,cdcl3)δ166.73,160.12,158.72,145.33,139.88,139.12,138.85,137.93,137.21,134.38,131.54,130.41,129.53,128.93,128.16,121.09,119.30,117.83,114.90,113.25,112.74,69.66,66.09,55.40.
[0122]
2、化合物3-6的制备
[0123][0124]
化合物3-6的合成方法与化合物1相似,ra1酰氯化后分别与rb4、rb5、rb6和rb10反应生成对应的(e)-3-(3-氟苯氧基)肉桂酸苄酯(化合物3)、(e)-3-(4-氟苯氧基)肉桂酸苄酯(化合物4)、(e)-3-(2-氟苯氧基)肉桂酸苄酯(化合物5)和(e)-3-(2,6-二氟苯氧基)肉桂酸苄酯(化合物6)。
[0125]
1)(e)-3-(3-氟苯氧基)肉桂酸苄酯(化合物3)的制备
[0126][0127]
合成步骤同化合物1,得到(e)-3-(3-氟苯氧基)肉桂酸苄酯(化合物3)(0.98g,两步产率96%),为白色固体。
[0128]1h nmr(400mhz,cdcl3)δ7.72(d,j=16.0hz,1h),7.52-7.41(m,2h),7.37-7.31(m,
4h),7.23(dd,j=15.3,7.7hz,1h),7.17(d,j=7.6hz,1h),7.09(s,1h),6.97(d,j=8.1hz,1h),6.76(t,j=7.1hz,2h),6.74
–
6.67(m,1h),6.47(d,j=16.0hz,1h),5.21(s,2h).
13
c nmr(100mhz,cdcl3)δ166.64,156.66,145.43,138.48,134.36,130.67,130.57,130.48,130.16,128.96,128.21,123.58,119.05,119.02,117.74,114.22,114.19,110.25,110.04,106.42,106.18,65.73.
[0129]
按照此方法也可获得(e)-3-(4-氟苯氧基)肉桂酸苄酯(化合物4)和(e)-3-(2-氟苯氧基)肉桂酸苄酯(化合物5)。
[0130]
2)(e)-3-(2,6-二氟苯氧基)肉桂酸苄酯(化合物6)的制备
[0131][0132]
合成步骤同化合物1,获得白色固体(e)-3-(2,6-二氟苯氧基)肉桂酸苄酯(化合物6)(0.97g,两步产率90%)。
[0133]1h nmr(400mhz,cdcl3)δ7.64(d,j=16.0hz,1h),7.44(d,j=3.0hz,2h),7.33
–
7.29(m,3h),7.17(d,j=4.6hz,1h),7.12(t,j=8.3hz,2h),6.99(s,1h),6.89
–
6.80(m,2h),6.65(d,j=8.3hz,1h),6.40(d,j=16.0hz,1h),5.15(s,2h).
19
f nmr(376mhz,cdcl3)δ-104.42.
13
cnmr(100mhz,cdcl3)δ166.79,156.73,145.59,138.54,134.42,130.59(2c),130.25(2c),129.07,128.80,128.70(2c),128.29(2c),123.62,118.27,117.77,115.35,111.66,111.44,65.79.
[0134]
3、化合物7-10的制备
[0135]
1)(e)-3-(4-(1-叔丁氧羰基-2,3,6-三氢吡啶基))肉桂酸苄酯(化合物7)和(e)-3-(4-(1-叔丁氧羰基哌啶基))肉桂酸苄酯(化合物9)的制备
[0136][0137]
化合物7和化合物9的合成步骤与化合物1相似,ra1酰氯化后分别与rb7、rb8反应生成对应的(e)-3-(4-(1-叔丁氧羰基-2,3,6-三氢吡啶基))肉桂酸苄酯(化合物7)和(e)-3-(4-(1-叔丁氧羰基哌啶基))肉桂酸苄酯(化合物9)。
[0138][0139]
合成步骤同化合物1,获得白色固体(e)-3-(4-(1-叔丁氧羰基-2,3,6-三氢吡啶基))肉桂酸苄酯(化合物7)(1.17g,两步产率95%)。
[0140]1h nmr(400mhz,cdcl3)δ7.72(d,j=16.0hz,1h),7.54
–
7.45(m,2h),7.42(s,1h),7.35-7.29(m,6h),6.48(d,j=16.0hz,1h),6.04(s,1h),5.24(s,2h),4.16
–
3.97(m,2h),3.62(t,j=5.8hz,2h),2.61
–
2.44(m,2h),1.50(s,9h).
13
c nmr(100mhz,cdcl3)δ166.57,154.70,145.11,144.85,142.24,141.00,136.17,134.22,130.27,129.32,128.79,128.60,128.02(2c),127.09,124.85,124.76,117.73,79.53,66.23,43.83,39.74,28.42(3c),27.32.
[0141]
2)(e)-3-(4-(1,2,3,6-四氢吡啶基))肉桂酸苄酯(化合物8)和(e)-3-(4-哌啶基)肉桂酸苄酯(化合物10)的制备
[0142][0143]
化合物8和化合物10的合成步骤相同,均为脱除n-boc保护基反应。
[0144][0145]
取化合物9(0.50g,1.19mmol)溶解于10ml dcm中,冰浴并缓慢加入三氟乙酸(1.33ml,17.91mmol),室温搅拌过夜,tlc监测,待反应完全后冰浴缓慢加入饱和nahco3溶液至ph为8左右,二氯甲烷/饱和食盐水体系萃取,收集有机层后加入过量无水硫酸钠干燥0.5h随后过滤并减压浓缩,经中性氧化铝柱色谱法纯化(dcm:(10甲醇:1氨水)=8:1,体积比),得到白色固体(e)-3-(4-哌啶基)肉桂酸苄酯(化合物10)(0.33g,87%)。
[0146]1h nmr(400mhz,cdcl3)δ7.69(d,j=15.4hz,1h),7.52(d,j=7.1hz,2h),7.35(d,j=7.9hz,3h),7.19(t,j=7.6hz,1h),7.08
–
6.89(m,4h),4.88(d,j=12.9hz,1h),4.22(d,j=13.2hz,1h),3.18(t,j=13.6hz,1h),2.84
–
2.63(m,2h),2.32(s,3h),1.91(d,j=13.2hz,2h),1.79
–
1.59(m,2h),1.27(d,j=10.0hz,1h).
13
c nmr(100mhz,cdcl3)δ165.43,145.16,142.49,138.12,135.44,129.51,128.79,128.49,127.74,127.57,127.27,123.78,117.63,46.66,43.10,42.86,34.15,32.99,29.71,29.67,21.49.
[0147]
4、化合物11的制备
[0148][0149]
合成步骤同化合物1,获得(3-苯氧基苄基)-3-苯基丙酸酯(化合物11)(0.95g,产率98%)。
[0150]1h nmr(400mhz,cdcl3)δ7.32(t,j=7.9hz,2h),7.29
–
7.21(m,3h),7.17(t,j=7.1hz,3h),7.11
–
7.10(m,1h),7.00(d,j=7.9hz,3h),6.97
–
6.90(m,2h),5.06(s,2h),2.95(t,j=7.8hz,2h),2.67(t,j=7.7hz,2h).
13
c nmr(100mhz,cdcl3)δ172.64,157.57,156.96,140.39,139.76,137.98,137.23,129.94,129.86,128.57,128.34,126.36,123.54,123.28(2c),122.71,119.12,118.41,118.29,65.81,35.91,30.98.
[0151]
5、化合物12和化合物13的制备
[0152]
1)(e)-(3-苯氧基)-4-甲氧基肉桂酸苄酯(化合物12)的制备
[0153][0154]
将间苯氧基苄醇(2.6ml,15mmol)放置于干燥的100ml圆底烧瓶中,加入超干二氯甲烷(20ml)使其完全溶解,0℃下依次滴加乙酸酐(1.7ml,18mmol)和无水三乙胺(6.3ml,45mmol),在室温下反应4h。tlc监测反应结束后,用dcm与饱和nacl溶液萃取,收集dcm层,加入无水na2so4干燥,过滤,收集滤液并浓缩。通过柱层析色谱法分离纯化(洗脱剂为石油醚:乙酸乙酯=10:1),减压浓缩,得到白色粉末状化合物12-1(3.18g,89%)。
[0155]
将化合物11-1(0.27g,1.11mmol),大茴香醛(0.10g,0.73mmol),三乙胺(0.3ml,2.22mmol)加入到50ml圆底烧瓶中,加入二氯甲烷使之溶解,在室温下反应40min后,加入四氯化钛(1.5ml,13.99mmol),室温继续反应5h。tlc监测反应结束后,用dcm与饱和nacl溶液萃取,收集dcm层,加入无水na2so4干燥,过滤,收集滤液并浓缩。通过柱层析色谱法分离纯化(洗脱剂为石油醚:乙酸乙酯=8:1),减压浓缩,得到白色粉末状(e)-(3-苯氧基)-4-甲氧基肉桂酸苄酯(化合物12)(0.23g,87%)。
[0156]1h nmr(400mhz,cdcl3)δ7.67(d,j=15.9hz,1h),7.46(d,j=7.9hz,2h),7.37
–
7.28(m,3h),7.14
–
7.09(m,2h),7.07(s,1h),7.02(d,j=8.2hz,2h),6.95(d,j=8.1hz,1h),6.89(d,j=7.9hz,2h),6.34(d,j=15.9hz,1h),5.20(s,2h),3.81(s,3h).
13
c nmr(100mhz,cdcl3)δ167.04,161.53,158.23,157.59,157.00,138.67,138.33,137.76,129.96(2c),129.87,127.09,123.51(2c),122.75,121.23,119.12,119.05,118.36,115.20,114.39,65.70,55.39.
[0157]
2)(e)-(3-苯氧基)-3,4-二甲氧基肉桂酸苄酯(化合物13)的制备
[0158][0159]
合成步骤同化合物12,获得(e)-(3-苯氧基)-3,4-二甲氧基肉桂酸苄酯(化合物13)(0.21g,产率为91%)。
[0160]1h nmr(400mhz,cdcl3)δ7.66(d,j=15.9hz,1h),7.34(d,j=6.8hz,3h),7.13
–
7.11(m,4h),7.06
–
7.02(m,2h),7.01(s,1h),6.95(d,j=8.1hz,1h),6.86(d,j=8.2hz,1h),6.35(d,j=15.9hz,1h),5.21(s,2h),3.90(s,6h).
13
c nmr(100mhz,cdcl3)δ166.88,157.56,156.97,151.27,149.27,145.22,138.24,129.92,129.81(2c),127.35,123.46,122.76,122.72,119.05(2c),118.35(2c),115.43,111.08,109.69,65.71,55.98,55.90.
[0161]
5、化合物14-16的制备
[0162]
1)(e)-2-(4-吡啶基)肉桂酸叔丁酯(化合物14)的制备
[0163][0164]
将4-溴吡啶(7.00g,44.60mmol),2-甲酰基苯硼酸(6.52g,43.47mmol),碳酸钠(7.63g,71.98mmol),四三苯基膦钯(2.69g,2.33mmol)加入到100ml双颈圆底烧瓶中,加入thf的水溶液(thf:h2o=4:1,20ml),在90℃下冷凝回流,反应过夜。tlc监测反应结束后,室温冷却,垫硅藻土过滤,用dcm与饱和nacl溶液萃取,收集dcm层,加入无水na2so4干燥,过滤,收集滤液并浓缩。通过柱层析色谱法分离纯化(洗脱剂为石油醚:乙酸乙酯=5:1),通过旋转蒸发仪进行减压浓缩,得到白色粉末状化合物14-1(6.92g,87%)。
[0165]
将化合物14-1(0.27g,1.47mmol),大茴香醛(0.10g,0.86mmol),三乙胺(0.3ml,2.22mmol)加入到50ml圆底烧瓶中,加入二氯甲烷使之溶解,在室温下反应40min后,加入四氯化钛(1.5ml,13.99mmol),室温继续反应5h。tlc监测反应结束后,用dcm与饱和nacl溶液萃取,收集dcm层,加入无水na2so4干燥,过滤,收集滤液并浓缩。通过柱层析色谱法分离纯化(洗脱剂为石油醚:乙酸乙酯=8:1),减压浓缩,得到白色粉末状化合物(e)-2-(4-吡啶基)肉桂酸叔丁酯(化合物14)(0.23g,95%)。
[0166]1h nmr(400mhz,cdcl3)δ8.67(s,2h),7.72(d,j=5.9hz,1h),7.56(d,j=15.8hz,1h),7.44(d,j=3.2hz,2h),7.34(s,1h),7.27(s,2h),6.36(d,j=15.9hz,1h),1.48(s,9h).
13
cnmr(100mhz,cdcl3)δ165.81,149.73,149.13,147.91,141.20,140.89,139.78,132.62,130.01,129.85,128.83,126.97,124.64,122.17,80.64,28.15(3c).
[0167]
2)(e)-2-(4-吡啶基)肉桂酸苄酯(化合物15)的制备
[0168][0169]
合成步骤同化合物14,获得(e)-2-(4-吡啶基)肉桂酸苄酯(化合物15)(0.26g,76%)。
[0170]1h nmr(400mhz,cdcl3)δ8.68(d,j=3.1hz,2h),7.73
–
7.69(m,2h),7.52
–
7.40(m,2h),7.34(d,j=5.1hz,5h),7.25(d,j=4.7hz,3h),6.46(d,j=15.9hz,1h),5.20(s,2h).
13
cnmr(100mhz,cdcl3)δ166.23,149.70,149.68,147.83,142.97,139.92,135.97,132.43,130.24,130.08,128.97,128.60,128.27(2c),128.13(2c),127.13,124.67,120.01,66.34,29.71.
[0171]
3)(e)-(3-苯氧基)-2-(4-吡啶基)肉桂酸苄酯(化合物16)的制备
[0172][0173]
合成步骤同化合物14,获得(e)-(3-苯氧基)-2-(4-吡啶基)肉桂酸苄酯(化合物16)(0.34g,76%)。
[0174]1h nmr(400mhz,cdcl3)δ8.67(d,j=5.9hz,2h),7.71
–
7.68(m,2h),7.47
–
7.44(m,2h),7.33
–
7.28(m,4h),7.27
–
7.25(m,2h),7.12
–
7.07(m,2h),7.01(d,j=7.8hz,3h),6.95
–
6.93(m,1h),6.46(d,j=15.9hz,1h),5.17(s,2h).
13
c nmr(100mhz,cdcl3)δ166.15,158.23,159.43,149.78(2c),147.77,143.14(2c),139.96,138.00,132.40,130.29,130.11(2c),129.94,128.97,127.17(2c),124.65,123.49,122.60,119.86,119.06(2c),118.38,118.27,65.84.
[0175]
实施例4 cck-8细胞活力检测实验
[0176]
初步活性筛选后选取化合物14进行后续活性检测实验。选用hct-8和hct116两种结直肠癌细胞系并采用cck-8试剂盒进行细胞活力检测实验,为探讨化合物14对hct-8和hct116的细胞活力及增殖的影响,用不同浓度的化合物14或5-氟脲嘧啶(5-fu,阳性药)作用于hct-8和hct116细胞24h,实验方法如下:
[0177]
1)复苏hct-8和hct116细胞并培养至三代以上,pbs冲洗细胞两次后,使用胰蛋白酶消化细胞并离心,随后进行细胞计数。
[0178]
2)将细胞以每孔约1x104个细胞的密度接种于96孔板上(100μl/孔),并将其放入培养箱中培养过夜。
[0179]
3)预培养结束后,向96孔板中加入不同浓度(0、10、20、30、40、50μm)的化合物14,孵育24h。
[0180]
4)共孵育结束后,向96孔板中加入cck-8溶液(10μl/孔)。
[0181]
5)将96孔板在培养箱中孵育1-4h。
[0182]
6)用酶标仪测定在450nm处的吸光度,并计算细胞活力,公式如下:
[0183]
细胞活力(%)=(加药od-空白od)/(对照od-空白od)
[0184]
加药od:含有细胞、cck-8和化合物34孔位的吸光度
[0185]
空白od:含有培养基、cck-8孔位的吸光度
[0186]
对照od:含有细胞、cck-8孔位的吸光度。
[0187]
表1-1试剂耗材及设备
[0188][0189]
实验结果如图1和图2所示,化合物14对结直肠癌细胞hct-8和hct116细胞的活力有抑制作用,呈浓度依赖性。我们观察到当给药浓度大于等于40μm时,给药24h后化合物18对hct-8和hct116细胞的存活率都有明显的抑制作用。例如,化合物14作用24h后,hct-8细胞活力仅为46.07%,hct116细胞活力仅为40.76%,与阳性对照药物5-氟尿嘧啶相比均有显著性差异。这些结果表明化合物14抑制了hct-8和hct116细胞的生长。
[0190]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。