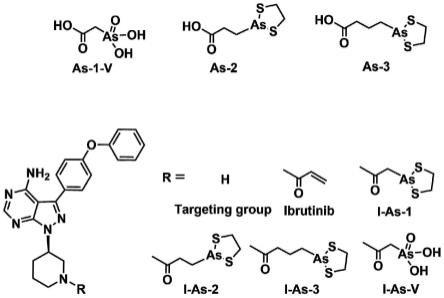
1.本发明属于生物医药技术领域,具体涉及一种共价靶向砷抑制剂及其制备方法和应用。
背景技术:
2.砷(arsenic,as)以其毒性广为人知,但另一方面,人类早在2000多年前就开始使用无机砷化合物(雄黄、雄黄和三氧化二砷等)治疗痈肿、癌变溃疡、癌症等疾病。福勒溶液(1%亚砷酸钾)是19世纪至20世纪初治疗梅毒、白血病、皮肤癌和其他疾病的常用药物。第一种现代意义上的化疗药物胂凡钠明也是一种砷药物。特别是砒霜(as2o3)以其对急性早幼粒细胞白血病(apl)的优异治疗效果而闻名于世。
3.砷致癌又能抗癌的性质反映了其独特的生物学特性,究其原因是砷化合物及其代谢产物通过与蛋白质半胱氨酸中的巯基结合改变蛋白质的构象和功能,影响砷结合蛋白的生理活动,导致砷致癌或抗癌。as2o3治疗apl的具体体机制是砷直接结合pml部分锌指基序中的半胱氨酸残基,改变pml的构象,诱导pml-rarα寡聚、泛素化,并在蛋白酶体降解,最终导致癌蛋白的降解和apl细胞凋亡。然而,as2o3的全身毒性和低生物利用度使得其对实体瘤和其他癌症的治疗效果并不显著。有机砷比无机砷更易于分子设计和化学修饰,是开发砷基药物的一个重要途径。具有不同化学结构的有机砷药物,如melarsoprol(美拉胂醇)、mer1(s-二甲基砷基硫代琥珀酸)、pazpao(对叠氮氧化苯砷)、gsao(4-(n-(s-谷胱甘肽乙酰基)氨基)苯基亚砷酸)和darinaparsin(二甲砷基谷胱甘肽)已被开发用于抗肿瘤或治疗其他疾病。然而,不断发展的小分子砷药物对癌细胞的毒性一直较低,其ic
50
仍处于微摩尔水平,而且砷和巯基之间的kd值在微摩尔水平,亲和力低,使现有的砷药物无法满足于临床用药需求。另一方面,现有的小分子砷药物没有特定的靶向官能团,会随机结合细胞中广泛存在的蛋白质半胱氨酸,而无法选择性地结合目标癌蛋白。因此,发展高选择性与特定癌蛋白高亲和力结合的共价靶向砷抑制剂的策略,不仅为开发新型砷基抗癌药物提供了新的方法和思路,也是极其必要的。
技术实现要素:
4.本发明目的在于克服现有技术缺陷,提供一种共价靶向砷抑制剂。
5.本发明的另一目的在于提供上述共价靶向砷抑制剂的制备方法。
6.本发明的再一目的在于提供上述共价靶向砷抑制剂的应用。
7.本发明的技术方案如下:
8.一种共价靶向砷抑制剂,其结构式为其中,r为
9.在本发明的一个优选实施方案中,所述r为
10.上述共价靶向砷抑制剂的制备方法,包括如下步骤:
11.(1)将有机砷配体、(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺、edc和nhs溶于dmf中,冰浴下加三乙胺调节ph为7.5-8.4,接着自然升温至室温后反应3-5h;该有机砷配体为砷乙酸、砷丙酸-乙二硫醇或砷丁酸-乙二硫醇;
[0012]
(2)将步骤(1)所得的物料用硅胶柱纯化,即得所述共价靶向砷抑制剂。
[0013]
在本发明的一个优选实施方案中,所述有机砷配体为砷乙酸,砷乙酸、(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺、edc和nhs的摩尔比为1∶0.25∶2∶1.2。
[0014]
在本发明的一个优选实施方案中,所述有机砷配体为砷丙酸-乙二硫醇,砷丙酸-乙二硫醇、(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺、edc和nhs的摩尔比为1∶1∶2∶1.2。
[0015]
在本发明的一个优选实施方案中,所述有机砷配体为砷丁酸-乙二硫醇,砷丁酸-乙二硫醇、(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺、edc和nhs的摩尔比为1∶1∶2∶1.2。
[0016]
在本发明的一个优选实施方案中,所述硅胶柱纯化中,正己烷和乙酸乙酯为流动相。
[0017]
上述共价靶向砷抑制剂在制备抗肿瘤药物中的应用。
[0018]
一种抗肿瘤药物,其有效成分包括上述共价靶向砷抑制剂。
[0019]
在本发明的一个优选实施方案中,其有效成分为上述的共价靶向砷抑制剂。
[0020]
本发明的有益效果是:本发明的共价靶向砷抑制剂具有靶向基团和三价砷(as
iii
)反应基团,该共价靶向砷抑制剂能够高度特异性地靶向btk,并高亲和力的共价结合btk半胱氨酸残基(cys481),有效抑制btk介导的b细胞受体(bcr)信号通路,导致ramos细胞死亡,具有较好的在体抗肿瘤增殖活性。
附图说明
[0021]
图1为本发明实施例1中的有机砷配体和共价靶向砷抑制剂的结构图。
[0022]
图2为本发明实施例2中的实验结果图,砷基化合物和ibrutinib对btk激酶活性抑制的量效曲线。
[0023]
图3为本发明实施例3中的实验结果图,通过竞争占位荧光标记评价共价靶向砷抑制剂与ramos细胞内btk的共价结合能力。pci-33380标记ramos细胞后,用凝胶电泳和凝胶荧光扫描检测探针标记的电泳结果图(约78kda,btk的分子量),条带经western blot检测证实是btk。
[0024]
图4为本发明实施例4中的i-as-1,i-as-2,i-as-3和i-as-v对ramos细胞内anti-igm刺激诱导的btk介导的bcr信号通路抑制的结果图,western blot实验使用相应的抗体
进行标记。
[0025]
图5为本发明实施例5中的通过凝胶电泳和凝胶荧光扫描对i-as-1和ibrutinib在ramos细胞内的蛋白质组反应性进行检测的结果图,箭头表示探针标记的主条带(约78kda,btk的预期分子量),该条带经western blot检测证实是btk。
[0026]
图6为本发明实施例6中的实验结果图,其中,(a)共价靶向砷抑制剂和ibrutinib对ramos细胞抗增殖活性的量效曲线,(b)ramos细胞对不同浓度i-as-1、i-as-2、i-as-3、i-as-v和ibrutinib的摄取结果图。
[0027]
图7为本发明实施例7中的i-as-1对小鼠移植瘤的抑制结果图,其中,(a)i-as-1在小鼠移植瘤模型实验中能更有效的阻止肿瘤生长,(b)在研究过程中,小鼠的体重随时间而变化,研究结束时每组小鼠的体重略有增加。
[0028]
图8为本发明实施例7中的i-as-1在小鼠心、肝、脾、肺、肾、肿瘤、血液中的生物分布结果图。
具体实施方式
[0029]
以下通过具体实施方式结合附图对本发明的技术方案进行进一步的说明和描述。
[0030]
本发明涉及的共价靶向砷抑制剂化学结构式为其中:
[0031]
当时,为(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷乙酸(i-as-v);
[0032]
当时,为(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷乙酸-乙二硫醇(i-as-1);
[0033]
当时,为(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷丙酸-乙二硫醇(i-as.2);
[0034]
当时,为(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷丁酸-乙二硫醇(i-as-3)。
[0035]
本发明的共价靶向砷抑制剂的制备方法的合成路线如图1所示:首先是制备羧基修饰的有机砷配体(砷乙酸/砷丙酸-乙二硫醇/砷丁酸-乙二硫醇),然后将有机砷配体与靶向基团(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺偶联,获得共价靶向砷抑制剂(图1),具体包括如下步骤:
[0036]
(1)合成有机砷配体
[0037]
a、合成砷乙酸(as-1-v):将1.2g(6mmol)三氧化二砷、1.44g(36mmol)氢氧化钠和112mg(0.6mmol)三甲基苄基氨溶于12ml水中,冰浴降温至20℃以下,往其中加入570mg
(6mmol)氯乙酸,室温反应5h。反应结束,加入2.16g(36mmol)冰醋酸析出未反应的无机砷,过滤去除沉淀,向滤液中接入2.16g(8.8mmol)二水合氯化钡,室温反应5h,生成白色沉淀,过滤,用冰水洗涤,将沉淀用聚苯乙烯磺酸氢离子型交换树脂(amberliter ir-120,h
)进行离子交换1h,旋除水得到白色固体产物砷乙酸(0.9g,81%)。1h nmr(500mhz,d2o)δ3.62(s,2h).
13
c nmr(126mhz,d2o)δ167.42,38.29.hrms(esi):m/z calcd for c2h5aso5[m h]
184.9426,found 184.9440.
[0038]
b、合成砷丙酸-乙二硫醇(as-2):将1.4g(7mmol)三氧化二砷和1.7g(42mmol)氢氧化钠加入到21ml水中,45℃搅拌溶解,滴加0.73g(7.7mmol)3-氯-1-丙醇,55℃反应4h。用浓盐酸调ph=2,浓缩至5ml,加入45ml乙醇除盐,旋除乙醇后得白色固体,不做进一步纯化,将所得固体与6.13g(28.7mmol)高碘酸钠和33.6mg三氯化钌加入到21ml水、14ml乙腈和14ml乙酸乙酯的混合溶液中,室温过夜反应。反应结束,加入25ml乙酸乙酯洗3遍,下层浊液加98ml水,用1m盐酸酸化得澄清溶液,加245ml乙醇提取,过滤得到的黄色澄清滤液,浓缩至70ml,加入5.26g(56mmol)1,2-乙二硫醇,室温反应2h。产物由反相色谱柱纯化,得到白色固体产物砷丙酸-乙二硫醇(0.65g,38.5%)1h nmr(600mhz,cdcl3)δ3.39-3.31(m,4h),2.67(t,j=7.6hz,2h),2.03(t,j=7.6hz,2h).
13
c nmr(151mhz,cdcl3)δ179.19,41.88,30.36,30.34.hrms(esi)m/z:calcd.for c5h9aso2s2[m h] :240.9333,found:240.9335.
[0039]
c、合成砷丁酸-乙二硫醇(as-3):0.2g(1mmol)三氧化二砷溶于3ml 10m氢氧化钠溶液中,1.08g(5mmol)二溴丁烷溶于0.5ml乙醇中,室温下将二溴丁烷溶液滴加到三氧化砷溶液中,80℃回流反应12h。用盐酸调节ph≈9,离心去除未反应的三氧化二砷,上清液减压旋蒸除水后用得到泛黄固体,不做进一步纯化,将所得固体与0.43g(2.1mmol)高碘酸钠和2.4mg三氯化钌用2.1ml水、1.4ml乙腈和1.4ml乙酸乙酯溶解,室温过夜反应。反应结束,加入3ml乙酸乙酯洗3遍,用1m盐酸酸化得澄清溶液,加20ml乙醇提取,过滤,滤液中加入0.38g(4mmol)1,2-乙二硫醇,室温反应2h。产物由反相色谱柱纯化,得到白色固体产物砷丁酸-乙二硫醇(95mg,37%)1h nmr(500mhz,chloroform-d)δ3.36-3.30(m,4h),2.45(t,j=6.9hz,2h),1.90-1.81(m,4h).
13
c nmr(126mhz,cdcl3)δ178.88,41.75,35.96,35.01,21.11.hrms(esi):m/z calcd for c6h
11
aso2s2[m h]
254.9489,found 254.9504.
[0040]
(2)将上述有机砷配体与(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺偶联,获得本发明的共价靶向砷抑制剂:
[0041]
a、合成(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷乙酸(i-as-v):100mg(0.54mmol)as-1-v,50mg(0.13mmol)(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺,207mg(1.08mmol)edc和75mg(0.65mmol)nhs溶于10ml dmf中,冰浴下加三乙胺调节ph=8,自然升温至室温反应4h。离心,所得上清液旋除溶剂后,用15ml纯水洗涤3次后,干燥得到白色固体产物(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷乙酸(43mg,61%)1h nmr(500mhz,dmso-d6)δ8.26(d,j=6.4hz,1h),7.67(dd,j=8.8,2.5hz,2h),7.52-7.38(m,2h),7.23-7.05(m,5h),4.87(m,0.5h),4.66(m,0.5h),4.56(br d,j=12.1hz,0.5h),4.25(br d,j=13.2hz,0.5h),4.13(br d,j=9.7hz,0.5h),3.97(br d,j=13.5hz,0.5h),3.75(m,0.5h),3.68(d,j=13.9hz,1h),3.50(d,j=14.2hz,1h),3.23-3.00(m,1h),2.87(m,0.5h),2.30-2.06(m,2h),1.94-1.73(m,2h).
13
c nmr(151mhz,dmso-d6)δ163.07,158.65,157.60,156.78,156.11,154.38,
143.89,143.67,130.61,128.36,124.26,119.44,97.82,55.39,53.04,52.49,50.66,46.93,46.08,42.07,30.10,29.96,24.86,23.75.hrms(esi):m/z calcd for c
24h25
asn6o5[m h]
553.1175,found:553.1181.
[0042]
b、合成(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷乙酸-乙二硫醇(i-as-1):185mg(1mmol)as-1-v、97mg(0.25mmol)(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺、382mg(2mmol)edc和138mg(1.2mmmol)nhs溶于25ml dmf中,冰浴下加三乙胺调节ph=8,自然升温至室温反应4h。离心,上清液中加入94.12mg(1mmol)1,2-乙二硫醇,室温反应2h,产物用硅胶柱纯化,用正己烷和乙酸乙酯为流动相(rf=0.25,正己烷∶乙酸乙酯∶=1∶1),得到白色固体(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷乙酸-乙二硫醇(116mg,78%)。
1
h nmr(500mhz,chloroform-d)δ8.26(d,j=20.4hz,1h),7.58(d,j=8.1hz,2h),7.46-7.36(m,2h),7.24-7.13(m,3h),7.09(d,j=8.0hz,2h),6.33(br d,j=24.4hz,2h),5.01-4.82(m,1h),4.74(br d,j=12.7hz,0.5h),4.55(br d,j=13.3hz,0.5h),4.07(br d,j=17.3hz,0.5h),3.87(br d,j=13.5hz,0.5h),3.72(m,0.5h),3.45-3.28(m,5h),3.08-2.92(m,2h),2.85(m,0.5h),2.42-2.20(m,2h),2.12-1.91(m,1h),1.82-1.66(m,1h).
13
c nmr(101mhz,cdcl3)δ169.31,163.89,159.76,155.75,153.67,151.56,145.89,130.15,129.78,124.58,119.92,119.26,97.09,54.49,53.45,50.75,46.72,45.65,42.32,42.26,41.38,41.13,29.97,25.02,23.88.hrms(esi):m/z calcd for c
26h27
asn6o2s2[m h]
595.0926,found:595.0949.
[0043]
c、合成(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷丙酸-乙二硫醇(i-as-2):48mg(0.2mmol)as-2、76mg(0.2mmol)(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺、77mg(0.4mmol)edc和28mg(0.24mmol)nhs溶于8ml dmf中,冰浴下加三乙胺调节ph=8,自然升温至室温反应4h。产物用硅胶柱纯化,用正己烷和乙酸乙酯为流动相(rf=0.3,正己烷:乙酸乙酯:=1:1),得到白色固体产物(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷丙酸-乙二硫醇(72mg,59%)。1h nmr(500mhz,chloroform-d)δ8.33(d,j=18.9hz,1h),7.64(dd,j=8.3,5.4hz,2h),7.43-7.36(m,2h),7.22-7.13(m,3h),7.09(d,j=7.9hz,2h),5.97(br s,2h),4.92-4.76(m,1.5h),4.51(br d,j=13.4hz,0.5h),4.03(br d,j=9.1hz,0.5h),3.87(br d,j=13.3hz,0.5h),3.70(m,0.5h),3.39-3.12(m,5h),2.88-2.71(m,2h),2.65(m,0.5h),2.46-2.20(m,2h),2.08-1.93(m,3h),1.78-1.65(m,1h).
13
c nmr(101mhz,cdcl3)δ171.39,158.75,157.73,156.25,155.17,154.10,144.29,130.03,127.38,124.18,119.63,119.17,98.55,53.38,52.56,49.87,45.95,45.59,42.16,41.61,31.99,30.23,29.85,25.51,25.04,23.89.hrms(esi):m/z calcd for c
27h29
asn6o2s2[m h]
609.1082,found:609.1117.
[0044]
d、合成(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷丁酸-乙二硫醇(i-as-3):51mg(0.2mmol)as-3、76mg(0.2mmol)(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-胺、76mg(0.4mmol)edc和28mg(0.24mmol)nhs溶于4ml dmf中,冰浴下加三乙胺调节ph约为8,自然升温至室温反应4h。产物用硅胶柱纯化,正己烷和乙酸乙酯为流动相(rf=0.36,正己烷∶乙酸乙酯:=1∶1),得到白色固体产物(r)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1h-吡唑并[3,4-d]嘧啶-4-砷丁酸-乙二硫醇(70mg,54%)。1h nmr(500mhz,chloroform-d)δ8.37(d,j=22.6hz,1h),7.66(dd,j=8.4,6.5hz,2h),7.45-7.38(m,2h),7.22-7.15(m,3h),7.11(d,j=7.9hz,2h),6.25-5.59(br s,2h),4.91-4.79(m,1.5h),4.58(br d,j=13.3hz,0.5h),4.05(br d,j=8.8hz,0.5h),3.88(br d,j=13.6hz,0.5h),3.75-3.64(m,1h),3.36-3.26(m,5h),3.16(m,0.5h),2.93-2.71(m,1.5h),2.53-2.21(m,5h),2.06-1.81(m,3h).
13
c nmr(101mhz,cdcl3)δ170.83,158.71,157.76,156.34,155.36,153.97,144.19,129.95,127.48,124.15,119.61,119.17,98.57,53.48,52.64,49.86,45.73,45.56,41.67,36.88,34.20,31.46,29.71,25.48,24.03,21.60,14.15.hrms(esi):m/z calcd for c
28h31
asn6o2s2[m h]
623.1239,found:623.1259.
[0045]
实施例2实施例1制备的共价靶向砷抑制剂在激酶分子水平对btk的抑制评价
[0046]
btk分子水平酶学分析采用kinase tk试剂盒测量各个化合物对btk的抑制效率,根据量效曲线(图2)确定各化合物对btk抑制的ic
50
值。结果显示,有机砷配体不能抑制btk的活性,共价靶向砷抑制剂和ibrutinib都能高效抑制btk的活性,ic
50
值在0.9nm-13.9nm(表1)。其中,三价砷药物i-as-1对btk的ic
50
为2.3nm,显著强于五价砷药物i-as-v,略强于三价砷药i-as-2和i-as-3。这表明,共价靶向砷抑制剂对btk有很高的亲和力,是靶向基团和三价砷基团协同作用实现与btk的高亲和力结合,并且碳链长度的短距离变化对共价靶向砷抑制剂和btk亲和力的影响很小。
[0047]
表1砷基化合物对btk激酶的ic
50
值
[0048][0049]
实施例3实施例1制备的共价靶向砷抑制剂与ramos细胞内btk的共价结合评价
[0050]
pci-33380探针是一种伊布替尼衍生物类btk共价结合探针,在表达btk的细胞中,可通过凝胶电泳和荧光凝胶扫描检测pci-33380与btk结合的荧光条带,并通过竞争标记评估btk抑制剂和btk之间的共价结合水平。ramos(6
×
106/2ml)细胞用pci-33380标记之前,先用不同浓度的共价靶向砷抑制剂孵育1h,用pbs洗3次洗去抑制剂,然后用pci-33380(2μm)标记1h。细胞用pbs洗3次去除多余的探针,用ripa(含磷酸酶抑制剂和蛋白酶抑制剂)裂解液裂解细胞,然后通过凝胶电泳和凝胶荧光扫描对细胞裂解液进行分析。如图3所示,i-as-1在60nm时就可以完全与ramos细胞内的btk共价结合,而靶向基团和i-as-v在35μm时仍不能与ramos细胞内的btk共价结合。这些结果进一步证明,共价靶向砷抑制剂对btk有很高
的亲和力,而且是靶向基团和三价砷基团起协作用使共价靶向砷抑制剂对btk具有高亲和力。
[0051]
实施例4实施例1制备的共价靶向砷抑制剂对btk介导的bcr信号通路的影响本实施例通过免疫印迹(western blot)研究了共价靶向砷抑制剂对ramos细胞内anti-igm刺激激活的btk介导的bcr信号通路的影响。ramos(6
×
106/2ml)细胞先用不同浓度的抑制剂预孵育1h,用pbs洗3次,然后用anti-igm(20μg/ml)刺激10min,细胞用pbs洗1次后用ripa(含磷酸酶抑制剂和蛋白酶抑制剂)裂解液裂解,细胞裂解液用凝胶电泳和western blot分析。结果如图4所示,三价砷药物i-as-1、i-as-2和i-as-3可以在纳摩尔水平上有效地靶向抑制bcr信号通路。i-as-1(64nm)显著阻断btk在y223处的自磷酸化、btk生理底物plcγ2(y1217)的磷酸化和下游激酶erk1/2(t202/y204)的磷酸化,但不影响btk上游激酶syk(y525/526)的磷酸化。五价砷抑制剂i-as-v在1μm时仍不能明显影响btk及其下游激酶的磷酸化。这些结果表明,i-as-1可以高选择性和高亲和力地抑制btk活性,从而抑制btk介导的bcr信号通路。
[0052]
实施例5实施例1制备的共价靶向砷抑制剂对ramos细胞内btk的选择性评价
[0053]
本实施例比较了i-as-1和ibrutinib在ramos细胞中蛋白质组的原位反应谱。ramos(6
×
106/2ml)细胞与不同浓度的i-as-1或ibrutinib(0.001-20μm)孵育1h,用pbs洗3次洗去抑制剂,然后用过高浓度的亲和探针pci-33380(20μm)标记1h,细胞用pbs洗3次去除多余的探针,用ripa(含磷酸酶抑制剂和蛋白酶抑制剂)裂解液裂解细胞,凝胶电泳和荧光凝胶扫描检测以显示脱靶荧光条带。本实施例观察到i-as-1在0.001-20μm范围内可以高选择性的对ramos细胞内的btk进行竞争标记,阻断btk的荧光标记,不影响其他脱靶荧光条带的标记,说明i-as-1对btk有很好的选择性(图5)。
[0054]
实施例6实施例1制备的共价靶向砷抑制剂对ramos细胞的抗增殖活性评价
[0055]
为了评价共价靶向砷抑制剂的抗肿瘤活性,本实施例评价了其对ramos细胞的抗增殖活性。rbmos细胞接种于96孔板(1
×
104cells/well/100μl)培养12h,然后用不同浓度的抑制剂处理72h(dmso,0.5%),用cck-8试剂盒测定细胞活力。根据量效曲线(图6a)确定抑制剂对ramos细胞的ic
50
值。结果很令人振奋,三价砷化合物i-as-1、i-as-2和i-as-3与ibrutinib相比,都表现出更强的抗ramos细胞增殖能力。特别是i-as-1抑制ramos增殖的ic
50
值为0.5μm,比ibrutinib提高了24倍,比五价砷i-as-v提高了200倍以上(表2)。本实施例通过细胞摄取实验分析这些结果可能的生物学基础。用不同浓度的i-as-1、i-as-2、i-as-3、i-as-v或ibrutinib培养ramos(5
×
106cells/2ml)细胞1h,然后用pbs洗3次,细胞提取液用hplc-icp-ms检测ramos细胞对含砷化合物的摄取量,用hplc-esi-ms检测ramos细胞对ibrutinib的摄取量。如图6b所示,在2μm-20μm范围内,i-as-1的摄取量分别是ibrutinib和i-as-v的6.3-13.8倍和19.0-54.8倍。其机制应该是i-as-1比i-as-v和ibrutinib的亲脂性更强,导致i-as-1更容易被ramos细胞摄取,这解释了i-as-1比ibrutinib和i-as-v具有更强细胞毒性。另一方面,本实施例注意到i-as-1的细胞毒性比i-as-2和i-as-3更强,但i-as-1的细胞摄取量略低于i-as-2和i-as-3。主要原因是i-as-1和btk之间的亲和力更高,更有利于与细胞内btk发生共价反应,抑制bcr信号通路杀死ramos细胞。总的来说,共价靶向砷抑制剂可以选择性高亲和力地靶向结合btk,抑制bcr信号通路,强效杀死ramos细胞。
[0056]
表2砷基抑制剂对ramos细胞的ic
50
值
[0057][0058]
实施例7实施例1制备的共价靶向砷抑制剂在体抗肿瘤活性评价
[0059]
为了进一步评估靶向btk的共价靶向砷抑制剂的抗肿瘤活性,本实施例用scid小鼠接种ramos细胞(5
×
106/0.1ml/只)构建小鼠移植瘤模型,用i-as-1和ibrutinib相对比来评价药物对小鼠移植瘤的抗肿瘤活性。当肿瘤体积可测量时,将小鼠随机分为四组(5只每组),分别尾静脉给药vehicle(75%生理盐水、5%dmso和20%hs 15)、i-as-1(10mg/kg、20mg/kg).和ibrutinib(10mg/kg)治疗14天,给药体积为0.1ml/d/只。如图7a所示,由于ramos细胞形成的小鼠异种移植瘤松散且不致密,当可以确认肿瘤形成时,肿瘤的初始大小较大,达到约300mm3。当治疗结束后,与未治疗组相比,ibrutinib治疗组(10mg/kg)的肿瘤体积只减小了15%(p=0.513),相同剂量i-as-1治疗组的肿瘤体积减小了17%(p=0.192)。令人惊喜的是,i-as-1(20mg/kg)治疗组的肿瘤体积减小了33%(p=0.02)。另一方面,ibrutinib给药15mg/kg后,小鼠出现死亡,因此未评估ibrutinib(20mg/kg)剂量组。治疗14天后,各组小鼠体重略有增加,治疗期间体重无明显波动,表明i-as-1毒性低,具有进一步开发应用的前景(图7b)。
[0060]
另一方面,砷是一种元素质量标签,可以通过icp-ms方便地检测砷药物在小鼠体内的生物分布情况。肿瘤治疗实验结束后,本实施例通过icp-ms检测小鼠心、肝、脾、肺、肾、肿瘤和血液中的砷含量,观察i-as-1在主要器官或组织中的砷累积情况。结果如图8所示,i-as-1在肿瘤组织中含量最多,有靶向肿瘤的倾向。
[0061]
综上所述,本发明设计并合成了具有高选择性和高亲和力共价结合癌蛋白btk的共价靶向砷抑制剂,抑制btk介导的bcr信号通路,高效抑制ramos细胞活性,并能在体抑制小鼠移植瘤的生长。结果表明,我们的策略为开发新型砷基抗肿瘤药物提供了清晰合理的设计思路和方案。
[0062]
以上所述,仅为本发明的较佳实施例而已,故不能依此限定本发明实施的范围,即依本发明专利范围及说明书内容所作的等效变化与修饰,皆应仍属本发明涵盖的范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。