1.本发明涉及高效氟吡甲禾灵制备领域,尤其是涉及一种高效氟吡甲禾灵的生产方法。
背景技术:
2.随着我国农业生产的不断发展,除草剂的使用比例逐年增加。高效氟吡甲禾灵是由美国陶氏益农公司开发的芳氧苯氧丙酸酯类除草剂。其化学名称:2-[4-(3-氯-5-三氟甲基-2-吡啶氧基)苯氧基]丙酸甲酯,又名高效盖草能。高效氟吡甲禾灵是内吸传导型除草剂,由植物叶片、茎秆和根系吸收,在植物体内抑制脂肪酸的生物合成,从而使植物细胞的生长、分裂停止,破坏细胞膜等含脂结构,最终导致杂草死亡。该除草剂经茎叶处理后,能够快速被禾本科类草的叶片吸收,并传导至整个植株,抑制植物分生组织而杀死禾草。同时,喷洒落入土壤中的高效氟吡甲禾灵也能够很容易的被根部吸收,也能起杀草作用,其在土壤中的半衰期平均为55天。与盖草能相比,高效盖草能在结构上以甲基取代盖草能中的乙氧乙基;并由于盖草能结构中丙酸的a-碳为不对称碳原子,故存在r和s两种光学异构体,其中s体没有除草活性,高效盖草能是除去了非活性部分(s体)的精制品(r体)。同等剂量下它比盖草能活性高,药效稳定,受低温、雨水等不利环境条件影响小。
[0003]
现有技术中,高效氟吡甲禾灵的合成方法主要为:先合成中间体2,3-二氯-5-(三氟甲基)吡啶,再合成高效氟吡甲禾灵,即2-[4-(3-氯-5-三氟甲基-2-吡啶氧基)苯氧基]丙酸甲酯。其中,中间体2,3-二氯-5-(三氟甲基)吡啶目前的合成方法主要有,1)采用2-氨基-5-甲基吡唑为原料,经溴化、氯化制得,但是该合成方法至少需进行三步反应,工艺流程长,且反应条件较为严苛,产物不易分离,不能适用于工业化生产。2)采用2-氨基-5-甲基吡唑为原料,与氯化剂反应后,经氟化制得,但是该合成方法的反应过程中,会发生较多的副反应,导致副产杂质较多,且分离较为困难,用于高效氟吡甲禾灵的制备中,无法保证最终产品的纯度。
[0004]
进一步的,发明人还发现,现有的采用2,3-二氯-5-(三氟甲基)吡啶为原料,合成高效氟吡甲禾灵工艺中,为保证良好的反应效果及产品纯度,往往需要较高反应温度(超过90℃),较长反应时间(约8h),生产能耗高,生产效率不高;并且,制得的高效氟吡甲禾灵纯度约95%,无法获得进一步提升。
技术实现要素:
[0005]
为解决现有技术中存在的技术问题,本发明提供一种高效氟吡甲禾灵的生产方法,能够克服现有高效氟吡甲禾灵合成技术的缺陷,能够在低反应温度、短反应时间条件下,获得良好的反应效果及产品纯度。
[0006]
为解决以上技术问题,本发明采取的技术方案如下:一种高效氟吡甲禾灵的生产方法,包括:制备中间体、氟化、缩合。
[0007]
所述制备中间体的方法为,将预定量的烟酸、三氯化磷投入至密闭反应容器内,搅
12:7-10:140-160。
[0011]
与现有技术相比,本发明的有益效果为:(1)本发明的高效氟吡甲禾灵的生产方法,通过在制备中间体步骤中设置第一至第三反应阶段,在不同温度、压力下进行阶段性反应,并于第四反应阶段中,设置负载有特定活性组分(氯化铁、氯化镧、六氯化钨)的催化剂,通过活性组分对反应的催化作用,能够对中间体2,3-二氯-5-(三氯甲基)吡啶的合成进行有效催化,减少中间体合成过程中副反应的发生,有效保证最终产品高效氟吡甲禾灵的反应效果及纯度指标;进一步的,在缩合步骤中,设置特定缚酸剂,并采用分次添加2,3-二氯-5-(三氟甲基)吡啶的方式,有效促进反应的进行,促进最终产物高效氟吡甲禾灵纯度及收率的同步提升;制得的高效氟吡甲禾灵纯度为99.234-99.260%(hplc面积归一法),收率为97.5-97.9%。
[0012]
(2)本发明的高效氟吡甲禾灵的生产方法,在以2,3-二氯-5-(三氟甲基)吡啶为原料,缩合制得高效氟吡甲禾灵的过程中,能够在较低反应温度(75-80℃),较短反应时间(5h)条件下,实现反应的顺利进行,有效实现高效氟吡甲禾灵纯度及收率的同步提升,其反应条件温和,反应效率高,有效降低生产能耗。
[0013]
(3)本发明的高效氟吡甲禾灵的生产方法,所采用的负载有活性成分的催化剂,即能够在制备中间体过程中进行有效催化,且所述催化剂可多次重复循环利用,进一步降低生产成本。
附图说明
[0014]
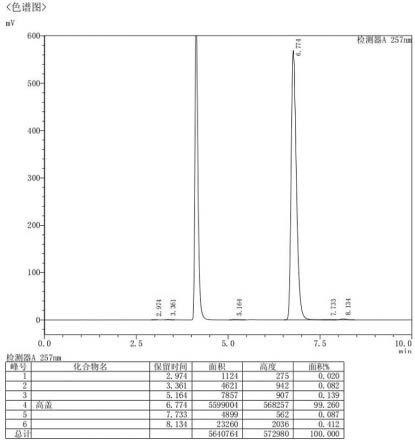
图1为实施例1制得的高效氟吡甲禾灵的液相色谱图。
[0015]
图2为实施例2制得的高效氟吡甲禾灵的液相色谱图。
[0016]
图3为实施例3制得的高效氟吡甲禾灵的液相色谱图。
具体实施方式
[0017]
为了对本发明的技术特征、目的和效果有更加清楚的理解,现说明本发明的具体实施方式。
[0018]
实施例1一种高效氟吡甲禾灵的生产方法,具体如下:1、制备催化剂:将预定量的氯化铁、氯化镧、六氯化钨投入至去离子水溶解完全,制得负载液;将活性炭投入至2倍体积的负载液中,30℃温度条件下,搅拌8h后,滤出固体物;固体物在真空环境下,65℃干燥至重量无变化,制得催化剂。
[0019]
其中,所述氯化铁、氯化镧、六氯化钨、去离子水的重量份比值为5:10:7:130。
[0020]
2、制备中间体:将预定量的烟酸、三氯化磷投入至密闭反应容器内,搅拌条件下,依次进行:1)第一反应阶段:以0.2℃/min的升温速率,升温至120℃,并加压至0.05mpa,保温保压1h;2)第二反应阶段:以3℃/min的升温速率,升温至150℃,并加压至0.1mpa,保温保压1h;
3)第三反应阶段:以10℃/min的升温速率,升温至200℃,保温7h;在第三反应阶段的保温过程中,维持压力在0.3-0.4mpa范围内。
[0021]
4)第四反应阶段:第三反应阶段制得的反应混合物自然冷却至常温,然后在105℃温度条件下,蒸馏至无气体馏出,将蒸馏剩余物置于装填有催化剂的密闭反应容器内,以0.2l/min的通气速率,通入氯气;通入完成后,升温至150℃,保温4h后,自然冷却至常温,滤除催化剂,然后在105℃温度条件下,保温至无气体馏出,制得中间体,即2,3-二氯-5-(三氯甲基)吡啶。
[0022]
其中,烟酸、三氯化磷、氯气的摩尔比为1:4.2:2.0。
[0023] 所述催化剂的装填量为反应容器内物料总质量的2wt%。
[0024]
3、氟化将制得的中间体2,3-二氯-5-(三氯甲基)吡啶、氯化铁、氟化氢投入至3倍体积的溶剂内,搅拌条件下,升温至150℃,保温反应12h;然后在搅拌条件下,自然冷却至115℃,保温搅拌2h后,自然冷却至常温;然后粗产物经二氯甲烷溶解,氢氧化钠水溶液洗涤,去离子水洗涤后,经蒸馏分离制得2,3-二氯-5-(三氟甲基)吡啶。
[0025]
其中,所述2,3-二氯-5-(三氯甲基)吡啶、氯化铁、氟化氢的重量份比值为100:2.5:50。
[0026]
所述溶剂为,浓度为70wt%的吡啶水溶液。
[0027]
所述氢氧化钠水溶液中,氢氧化钠的浓度为0.8mol/l。
[0028]
4、缩合将(r)-( )-2-(4-羟基苯氧基)丙酸甲酯投入至密闭环境内,采用氮气置换三次后,依次投入n,n-二甲基乙酰胺、缚酸剂,搅拌均匀后,升温至70℃保温;继续投入70%剂量的2,3-二氯-5-(三氟甲基)吡啶,搅拌条件下,升温至75℃,保温反应2h后;继续投入剩余剂量的2,3-二氯-5-(三氟甲基)吡啶,搅拌条件下,升温至80℃,保温反应3h,自然冷却至常温,滤除固体物;将制得的滤液置于真空环境下脱除溶剂后,投入至2倍体积的正己烷中,搅拌均匀后,采用去离子水水洗2次;置于真空环境下,蒸馏脱除正己烷后,升温至115℃,继续保温蒸馏至无气体馏出,制得高效氟吡甲禾灵。
[0029]
其中,(r)-( )-2-(4-羟基苯氧基)丙酸甲酯、n,n-二甲基乙酰胺、缚酸剂、2,3-二氯-5-(三氟甲基)吡啶的重量份比值为10:45:6:12。
[0030]
所述缚酸剂,为三乙胺和醋酸钠。所述三乙胺和醋酸钠的重量份比值为3:1。
[0031]
经检测,本实施例制得的高效氟吡甲禾灵的纯度为99.260%(hplc面积归一法),收率为97.5%,具体液相色谱图见附图1。
[0032]
实施例2一种高效氟吡甲禾灵的生产方法,包括:制备催化剂、制备中间体、氟化、缩合。
[0033]
1、制备催化剂:将预定量的氯化铁、氯化镧、六氯化钨投入至去离子水溶解完全,制得负载液;将活性炭投入至3倍体积的负载液中,35℃温度条件下,搅拌10h后,滤出固体物;固体物在真空环境下,70℃干燥至重量无变化,制得催化剂。
[0034]
其中,所述氯化铁、氯化镧、六氯化钨、去离子水的重量份比值为7:11:8:150。
[0035]
2、制备中间体:
将预定量的烟酸、三氯化磷投入至密闭反应容器内,搅拌条件下,依次进行:1)第一反应阶段:以0.3℃/min的升温速率,升温至125℃,并加压至0.08mpa,保温保压1.5h;2)第二反应阶段:以4℃/min的升温速率,升温至155℃,并加压至0.15mpa,保温保压1.5h;3)第三反应阶段:以12℃/min的升温速率,升温至205℃,保温8h;在第三反应阶段的保温过程中,维持压力在0.3-0.4mpa范围内。
[0036]
4)第四反应阶段:第三反应阶段制得的反应混合物自然冷却至常温,然后在110℃温度条件下,蒸馏至无气体馏出,将蒸馏剩余物置于装填有催化剂的密闭反应容器内,以0.4l/min的通气速率,通入氯气;通入完成后,升温至155℃,保温5h后,自然冷却至常温,滤除催化剂,然后在110℃温度条件下,保温至无气体馏出,制得中间体,即2,3-二氯-5-(三氯甲基)吡啶。
[0037]
其中,烟酸、三氯化磷、氯气的摩尔比为1:4.3:2.1。
[0038] 所述催化剂的装填量为反应容器内物料总质量的3wt%。
[0039]
3、氟化将制得的中间体2,3-二氯-5-(三氯甲基)吡啶、氯化铁、氟化氢投入至4倍体积的溶剂内,搅拌条件下,升温至155℃,保温反应14h;然后在搅拌条件下,自然冷却至120℃,保温搅拌2.5h后,自然冷却至常温;然后粗产物经二氯甲烷溶解,氢氧化钠水溶液洗涤,去离子水洗涤后,经蒸馏分离制得2,3-二氯-5-(三氟甲基)吡啶。
[0040]
其中,所述2,3-二氯-5-(三氯甲基)吡啶、氯化铁、氟化氢的重量份比值为100:2.7:52。
[0041]
所述溶剂为,浓度为72wt%的吡啶水溶液。
[0042]
所述氢氧化钠水溶液中,氢氧化钠的浓度为1mol/l。
[0043]
4、缩合将(r)-( )-2-(4-羟基苯氧基)丙酸甲酯投入至密闭环境内,采用氮气置换三次后,依次投入n,n-二甲基乙酰胺、缚酸剂,搅拌均匀后,升温至73℃保温;继续投入75%剂量的2,3-二氯-5-(三氟甲基)吡啶,搅拌条件下,升温至78℃,保温反应2.5h后;继续投入剩余剂量的2,3-二氯-5-(三氟甲基)吡啶,搅拌条件下,升温至83℃,保温反应2.5h,自然冷却至常温,滤除固体物;将制得的滤液置于真空环境下脱除溶剂后,投入至2.5倍体积的正己烷中,搅拌均匀后,采用去离子水水洗2次;置于真空环境下,蒸馏脱除正己烷后,升温至120℃,继续保温蒸馏至无气体馏出,制得高效氟吡甲禾灵。
[0044]
其中,(r)-( )-2-(4-羟基苯氧基)丙酸甲酯、n,n-二甲基乙酰胺、缚酸剂、2,3-二氯-5-(三氟甲基)吡啶的重量份比值为10:48:6.2:12.3。
[0045]
所述缚酸剂,为三乙胺和醋酸钠。所述三乙胺和醋酸钠的重量份比值为3.5:1。
[0046]
经检测,本实施例制得的高效氟吡甲禾灵纯度为99.234%(hplc面积归一法),收率为97.9%,具体液相色谱图见附图2。
[0047]
实施例3一种高效氟吡甲禾灵的生产方法,包括:制备催化剂、制备中间体、氟化、缩合。
[0048]
1、制备催化剂:
将预定量的氯化铁、氯化镧、六氯化钨投入至去离子水溶解完全,制得负载液;将活性炭投入至5倍体积的负载液中,40℃温度条件下,搅拌12h后,滤出固体物;固体物在真空环境下,75℃干燥至重量无变化,制得催化剂。
[0049]
其中,所述氯化铁、氯化镧、六氯化钨、去离子水的重量份比值为8:12:10:160。
[0050]
2、制备中间体:将预定量的烟酸、三氯化磷投入至密闭反应容器内,搅拌条件下,依次进行:1)第一反应阶段:以0.5℃/min的升温速率,升温至130℃,并加压至0.1mpa,保温保压2h;2)第二反应阶段:以5℃/min的升温速率,升温至160℃,并加压至0.2mpa,保温保压2h;3)第三反应阶段:以15℃/min的升温速率,升温至210℃,保温9h;在第三反应阶段的保温过程中,维持压力在0.3-0.4mpa范围内。
[0051]
4)第四反应阶段:第三反应阶段制得的反应混合物自然冷却至常温,然后在115℃温度条件下,蒸馏至无气体馏出,将蒸馏剩余物置于装填有催化剂的密闭反应容器内,以0.5l/min的通气速率,通入氯气;通入完成后,升温至160℃,保温6h后,自然冷却至常温,滤除催化剂,然后在115℃温度条件下,保温至无气体馏出,制得中间体,即2,3-二氯-5-(三氯甲基)吡啶。
[0052]
其中,烟酸、三氯化磷、氯气的摩尔比为1:4.5:2.2。
[0053] 所述催化剂的装填量为反应容器内物料总质量的4wt%。
[0054]
3、氟化将制得的中间体2,3-二氯-5-(三氯甲基)吡啶、氯化铁、氟化氢投入至6倍体积的溶剂内,搅拌条件下,升温至160℃,保温反应16h;然后在搅拌条件下,自然冷却至125℃,保温搅拌3h后,自然冷却至常温;然后粗产物经二氯甲烷溶解,氢氧化钠水溶液洗涤,去离子水洗涤后,经蒸馏分离制得2,3-二氯-5-(三氟甲基)吡啶。
[0055]
其中,所述2,3-二氯-5-(三氯甲基)吡啶、氯化铁、氟化氢的重量份比值为100:3:55。
[0056]
所述溶剂为,浓度为75wt%的吡啶水溶液。
[0057]
所述氢氧化钠水溶液中,氢氧化钠的浓度为1.2mol/l。
[0058]
4、缩合将(r)-( )-2-(4-羟基苯氧基)丙酸甲酯投入至密闭环境内,采用氮气置换三次后,依次投入n,n-二甲基乙酰胺、缚酸剂,搅拌均匀后,升温至75℃保温;继续投入80%剂量的2,3-二氯-5-(三氟甲基)吡啶,搅拌条件下,升温至80℃,保温反应3h后;继续投入剩余剂量的2,3-二氯-5-(三氟甲基)吡啶,搅拌条件下,升温至85℃,保温反应2h,自然冷却至常温,滤除固体物;将制得的滤液置于真空环境下脱除溶剂后,投入至3倍体积的正己烷中,搅拌均匀后,采用去离子水水洗2次;置于真空环境下,蒸馏脱除正己烷后,升温至125℃,继续保温蒸馏至无气体馏出,制得高效氟吡甲禾灵。
[0059]
其中,(r)-( )-2-(4-羟基苯氧基)丙酸甲酯、n,n-二甲基乙酰胺、缚酸剂、2,3-二氯-5-(三氟甲基)吡啶的重量份比值为10:50:6.5:12.5。
[0060]
所述缚酸剂,为三乙胺和醋酸钠。所述三乙胺和醋酸钠的重量份比值为4:1。
[0061]
经检测,本实施例制得的高效氟吡甲禾灵纯度为99.236%(hplc面积归一法),收率为97.6%,具体液相色谱图见附图3。
[0062]
对比例1采用实施例2的技术方案,其区别之处在于:省略制备催化剂步骤,将氯化铁、六氯化钨作为催化剂,直接用于制备中间体步骤;其中,氯化铁和六氯化钨的重量份比值为2:3。
[0063]
对比例1制得的高效氟吡甲禾灵的纯度为91.1%,收率为88.4%可以看出,本发明采用负载有氯化铁、氯化镧、六氯化钨的活性炭作为催化剂,并应用于第四反应阶段,能够对中间体2,3-二氯-5-(三氯甲基)吡啶的合成进行有效催化,减少副反应的发生,制得的中间体2,3-二氯-5-(三氯甲基)吡啶用于高效氟吡甲禾灵的合成中,能够有效保证其纯度指标。
[0064]
对比例2采用实施例2的技术方案,其区别之处在于:1)制备中间体步骤中,省略第一反应阶段至第三反应阶段,改为在烟酸、三氯化磷投料完成后,升温至205℃,保温反应8h,然后进行自然冷却、蒸馏等后续步骤;2)缩合步骤中,在n,n-二甲基乙酰胺、缚酸剂投入完成后,搅拌均匀,升温至70-75℃保温;然后投入全部剂量的2,3-二氯-5-(三氟甲基)吡啶,升温至80-85℃,保温反应5h,然后进行自然冷却、滤除固体物等后续步骤;其中,缚酸剂为碳酸钾。
[0065]
对比例2制得的高效氟吡甲禾灵的纯度为92.3%,收率为89.2%。
[0066]
可以看出,本发明在制备中间体步骤中,采用特定的第一反应阶段至第三反应阶段;以及在缩合步骤中,采用特定缚酸剂,并采用分次添加2,3-二氯-5-(三氟甲基)吡啶的方式,有效促进反应的进行,促进最终产物高效氟吡甲禾灵纯度及收率的同步提升。
[0067]
除非另有说明,本发明中所采用的百分数均为质量百分数。
[0068]
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。