1.本发明涉及螺环二酚骨架4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢茚-7,7
’‑
二酚和4, 4
’‑
二甲氧基-6,6
’‑
二叔丁基-1,1
’‑
螺二氢茚-7,7
’‑
二酚及其双亚膦酸酯化合物的制备方法和应用。
背景技术:
2.轴对称有机化合物一直是不对称催化领域的研究热点,轴对称化合物在生物医药、工业催化、功能材料等领域有着广泛的应用。已近成功工业化的binol、 binap等联芳基配体已经有了很广泛的应用。
[0003][0004]
1999年,birman等从丙酮和3-甲氧基苯甲醛出发,经过六步反应得到了外消旋的螺二氢茚二酚((
±
)-spinol)。该二酚与氯甲酸薄荷醇酯形成的非对映异构体可以通过柱层析进行分离,从而得到光学纯的(r)-( )-spinol和 (s)-(-)-spinol。us20130135574a1、cn1055003542a也报道了类似的合成路线和拆分方法。在此基础上,南开大学的周其林等在2002年报道了更为实用的拆分方法,他们利用氯化苄基辛可宁啶和对映异构体中的一种易形成包结物的特点,通过简单的回流、冷却、结晶、过滤和酸化等步骤就可以得到光学纯的螺二氢茚二酚。2016年谭斌等人报道了手性膦酸催化下spinol的不对称合成,直接从 1,5-双(5-羟基-2-甲基苯基)-3-戊酮一步环化脱水到(s)-4,4
’‑
二甲基-7,7
’‑
二羟基
ꢀ‑
1,1
’‑
螺二氢茚(收率97%,ee值90%)。值得一提的事,他们所使用的配体是以手性spinol为骨架的膦酸。此外,cn109761774a研究了一种从1,5-双(3
‑ꢀ
羟基苯基)-3-戊酮傅克环化到消旋spinol方法,这是首例有关羟基对位不需要占位基团即可环化合成1,1
’‑
螺二氢茚-7,7
’‑
二酚的报道。
[0005]
氢甲酰化反应自1938年被otto roelen教授发现以来,在工业当中得到了非常巨大的应用。以丙烯为原料氢甲酰化生产丁醛,经缩合加氢后生产丁辛醇(主要用于生产邻苯二甲酸二辛酯即dop)的生产和消费量最大,其国内每年需求超过300万吨。由于dop分子量小且易挥发,基于环保和健康安全考虑,全球趋势是使用更高分子量、低挥发性和更加稳定的增塑剂。而丁烯氢甲酰化生产戊醛,经缩合加氢制得2-丙基庚醇(2-ph),由2-ph生产的dphp增塑剂能很好地解决dop所面临的环保、健康、安全方面的担忧。迄今为止,美国和欧盟等国已开始使用dphp替代dop,这一趋势已开始影响亚洲市场。
[0006]
混合碳四氢甲酰化法是目前报道的戊醛生产工艺中公认的最经济、最直接的路线。美国联合碳化公司(现陶氏化学)开发的联苯双亚膦酸酯型配体(biphephos) 与铑组成的催化体系可以使混合碳四中的2-丁烯有效进行氢甲酰化反应且可获得高的正异比。例如
us4668651、us4769498、us4148830、cn86106770和 cn86106811中提出的双亚膦酸酯配体可使其正异比达到26。而基于biphephos 的第四代双亚膦酸酯催化的氢甲酰化生产工艺已经实现工业化。
[0007]
在氢甲酰化反应中,带有联苯、联萘和蒽骨架的双齿、多齿亚膦酸酯/亚膦酰胺配体(如:biphephos、anthracenetriol-based triphosphite)被国外大型化学公司如巴斯夫、陶氏化学和赢创以及一些研究小组广泛报导及专利化,而以螺环双亚膦酸酯/亚膦酰胺配体的报道非常少。2012年,丁奎琳等人报道一系列螺缩酮双亚膦酰胺配体,在1-己烯和其它端烯的氢甲酰化反应中表现出优异的转化率 (90%)和正异比(l/b=174.4),但在催化内烯烃(如:反-2-丁烯)的氢甲酰化反应的转化率仅有不到15%。因此,开发更高效、更高选择性和稳定性的新型螺环双膦配体具有重要意义和工业化应用价值。
[0008][0009]
本发明中发展的螺环二酚及其双亚膦酸酯配体的新型制备方法,从起始原料到消旋的螺二酚仅四步反应,具有易于合成、适合放大合成、无昂贵试剂和金属催化剂、可以产业化等特点。该工艺路线简单,四步收率70%、避免了剧烈和危险的实验条件和试剂、所以原料均可回收利用等特点。此外,新型螺环双亚膦酸酯配体(消旋体)可用于催化以廉价的醚后碳四或mto碳四为原料的氢甲酰化反应。
技术实现要素:
[0010]
本发明实施例的目的是在于提供螺环二酚及其双亚膦酸酯制备方法和应用。
[0011]
本发明的实施例是这样实现的,螺环二酚及其双亚膦酸酯化合物,其结构如通式i所示:
[0012][0013]
其中,通式i及其衍生物的结构表示如下:
[0014][0015]
本发明实施例的另一目的在于提供螺环二酚及其双亚膦酸酯化合物的制备方法和应用,所述螺环双亚膦酸酯化合物是以4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢茚-7,7
’‑
二酚化合物或4,4
’‑
二甲氧基-6,6
’‑
二叔丁基-1,1
’‑
螺二氢茚-7,7
’‑
二酚为原料,在有机溶剂以及正丁基锂或三乙胺的作用下与含有芳基或环状芳基结构的氯代亚膦酸酯反应而成;所述螺环双亚膦酸酯配体为l1-l31中的一种。
附图说明
[0016]
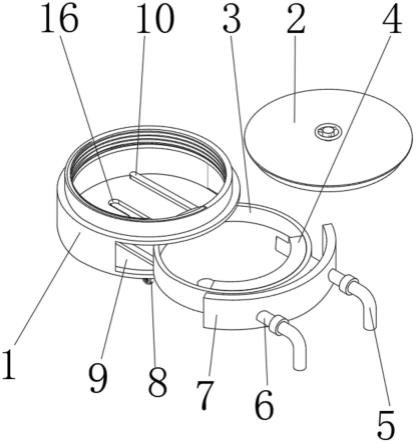
图1是本发明对比实施例中所使用的间歇式烯烃小试评价装置。
具体实施方式
[0017]
下面通过实施例对本发明的以上路线进行具体的描述,为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用于解释本发明,并不用于限定本发明。
[0018]
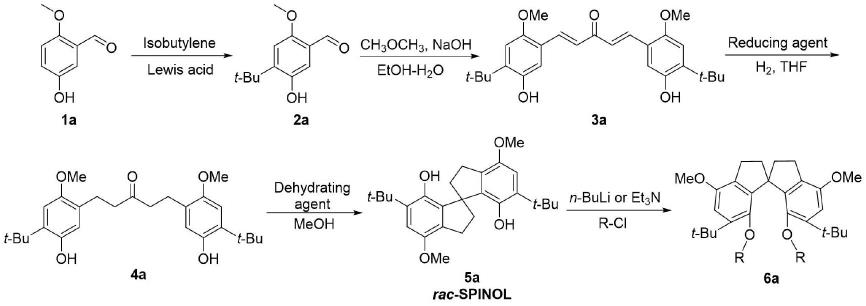
本发明公开提供了两种螺二酚骨架的合成路线,分别以3-羟基苯甲醛或5-羟基-2-甲氧基苯甲醛出发,经过烷基化、羟醛缩合、氢化还原、环化脱水后得到对应的螺环二酚,再与氯代亚膦酸酯经酯化反应后得到螺环双亚膦酸酯配体。
[0019]
具体地说,本发明所涉及的合成方法具体说明如下:
[0020]
在一些实施方式中,3-羟基苯甲醛或5-羟基-2-甲氧基苯甲醛与异丁烯在路易斯酸催化下发生烷基化反应得到2,4-二叔丁基-5-羟基苯甲醛(2)或2-甲氧基-4
‑ꢀ
叔丁基-5-羟基苯甲醛(2a)。
[0021]
在一些实施方式中,所述烷基化反应所用的质子酸或路易斯酸为有机酸或无机酸中的一种或多种,有机酸如:甲酸、乙酸、乙二酸、二氯乙酸、三氟乙酸、丙酸、丙二酸、丙酮酸、丁酸、戊酸、己酸、己二酸、苯甲酸、对硝基苯甲酸、对苯二甲酸、苯磺酸、氟磺酸、甲磺酸、三氟甲磺酸、对甲苯磺酸等;无机酸如:氢溴酸、盐酸、氢氟酸、亚硫酸、硫酸、高氯酸、膦酸、焦磷酸、硝酸、亚硝酸、铬酸、氟锑磺酸、氟锑酸等;烷基化试剂为溴代叔丁烷、氯代叔丁烷、异丁烯、叔丁醇中的任意一种;反应温度为80~140℃,反应溶剂为苯、甲苯、对甲苯、对二甲苯、邻二甲苯、氯苯或二氯苯中的任意一种。
[0022]
在一些实施方式中,中间体2或2a与丙酮经羟醛缩合反应得到1,5-双(2,4
‑ꢀ
二叔丁基-5-羟基苯基)-1,4-戊二烯-3-酮(3)或1,5-双(2-甲氧基-4-叔丁基-5-羟基苯基)-1,4-戊二烯-3-酮(3a)。
[0023]
在一些实施方式中,中间体2或2a与丙酮缩合反应在碱的存在下,混合后在溶剂中进行反应。
[0024]
在一些更优选的实施方式中,所述碱是氢氧化钾、氢氧化钠、碳酸钾、碳酸钠、叔丁醇钠、叔丁醇钾中的任意一种;碱的用量1.5~10当量,所述溶剂是乙醇、水或其混合溶剂,混合溶剂比例介于50:50和90:10之间,反应温度为20 ~75℃。
[0025]
在一些实施方式中,中间体3或3a在金属催化剂的催化下加氢还原碳碳双键,得到1,5-双(2,4-二叔丁基-5-羟基苯基)-3-戊酮(4)或1,5-双(2-甲氧基-4-叔丁基
ꢀ‑
5-羟基苯基)-3-戊酮(4a)。
[0026]
在一些实施方式中,所述还原反应的催化剂为雷尼镍、氯化铁、氧化钴或钯碳中的任意一种;所述还原催化剂的用量催化剂的用量为1~20%(w/w),氢气压力0.05~5mpa,反应温度为20~50℃,反应时间为24~72个小时。反应溶剂为乙酸乙酯、四氢呋喃、二氯甲烷或1,4-二氧六环中的任意一种。
[0027]
在一些实施方式中,中间体4或4a在脱水剂的作用下经环化反应得到4,4’,6, 6
’‑
四叔丁基-1,1
’‑
螺二氢茚-7,7
’‑
二酚(5)或4,4
’‑
二甲氧基-6,6
’‑
二叔丁基-1,1
’‑ꢀ
螺二氢茚-7,7
’‑
二酚(5a)。
[0028]
在一些实施方式中,所述脱水剂为多聚磷酸、浓硫酸、乙酸酐、甲磺酸、苯甲酸、对甲苯磺酸、无水三氯化铝中的任意一种。反应溶剂为甲苯、正庚烷、二氯甲烷、三氯甲烷、二氯乙烷中的任意一种。脱水剂的用量为10~70当量,反应温度为45~135℃,反应时间为2~
6个小时。
[0029]
在一些实施方式中,中间体5或5a与正丁基锂反应得到锂化后反应液;锂化后反应液与含有芳基或环状芳基结构的氯代亚膦酸酯反应,得到大位阻螺环双亚膦酸酯化合物。
[0030]
在一些实施方式中,中间体5或5a与含有芳基或环状芳基结构的氯代亚膦酸酯和缚酸剂的混合溶液反应,得到大位阻螺环双亚膦酸酯化合物。
[0031]
在一些实施方式中,所述酯化反应中,正丁基锂的用量2~4当量;缚酸剂为三乙胺、n,n-二异丙基乙胺、吡啶中的任意一种,用量为5~20当量;反应温度为-78~80℃,反应时间12~48小时,反应溶剂为甲苯、四氢呋喃、乙醚、2
‑ꢀ
甲基四氢呋喃、甲基叔丁基醚、异丙醚、苯甲醚、乙二醇二甲醚、二乙二醇二甲醚、丁醚、环戊基甲醚或1,4-二氧六环中的任意一种。
[0032]
实施例1:2,4-二叔丁基-5-羟基苯甲醛的制备(2)
[0033][0034]
在2l的双口瓶加入1(20.0g,163.8mmol),将反应瓶置换为氮气氛围,于 25℃下加入200ml四氢呋喃、浓硫酸(2.5g)。持续通入1.5个大气压力的异丁烯,加热至100℃反应持续12个小时。反应液经水淬灭后,加入300ml水并用乙酸乙酯萃取三次(每次300ml)。所得有机相经无水硫酸钠干燥后减压旋干得到淡黄色固体35.7g,收率94%。1h nmr(400mhz,cdcl3):δ=1.35(s,9h),1.41 (s,9h),6.54(s,1h),7.30(d,1h),7.38(s,1h),9.89(d,1h)。
[0035]
实施例2:2-甲氧基-4-叔丁基-5-羟基苯甲醛(2a)
[0036][0037]
中间体2a的合成步骤与中间体2相似,中间体1a投料20g,最终得到黄色固体26.3g,收率96%。1h nmr(400mhz,cdcl3):δ=1.39(s,9h),3.89(s,3h), 6.62(s,1h),6.96(s,1h),7.36(d,1h)。
[0038]
实施例3:1,5-双(2,4-二叔丁基-5-羟基苯基)-1,4-戊二烯-3-酮(3)
[0039][0040]
在500ml的双口瓶加入1(20.0g,85.3mmol)、丙酮(3.1ml,42.7mmol) 和125ml乙醇并形成混合溶液。随后,将混合溶液转移至200ml的滴液漏斗中,缓慢滴入400ml氢氧化钠的乙醇水溶液(19.0g naoh,etoh-h2o:65%)。滴加完后,在室温的中搅拌2小时,有机相用二氯甲烷稀释,用水洗涤,无水硫酸钠干燥,并通过快速柱层析分离,即得淡黄色油状目标产物19.3g,静置凝固,收率92%。1h nmr(400mhz,cdcl3):δ=1.31(s,18h),1.38(s,18h),6.60
(s,2h), 6.70
–
6.83(m,4h),7.35(s,2h),7.75(d,2h)。
[0041]
实施例4:1,5-双(2-甲氧基-4-叔丁基-5-羟基苯基)-1,4-戊二烯-3-酮(3a)
[0042][0043]
中间体3a的合成步骤与中间体3相似,中间体2a投料20g,最终得到黄色油状目标产物19.6g,收率95%。1h nmr(400mhz,cdcl3):δ=1.43(s,18h),3.88 (s,6h),6.69
–
7.01(m,8h),7.82(d,2h)。
[0044]
实施例5:1,5-双(2,4-二叔丁基-5-羟基苯基)-3-戊酮(4)
[0045][0046]
在500ml的圆底瓶中,依次加入3(15.0g,30.6mmol),200ml丙酮和60g 雷尼镍。然后,插入氢气球并在氢气氛围下搅拌反应,通过tlc监测反应进程。当原料点消失(反应约1天),滤出催化剂,用丙酮洗涤,滤液减压干后得到无色油状目标产物14.97g,收率99%。1h nmr(400mhz,cdcl3):δ=1.36(d,36h), 2.71
–
3.03(m,8h),6.07(s,2h),6.54(t,2h),7.15(s,2h)。
[0047]
实施例6:1,5-双(2-甲氧基-4-叔丁基-5-羟基苯基)-3-戊酮(4a)
[0048][0049]
中间体4a的合成步骤与中间体4相似,中间体3a投料15g,最终得到黄色油状目标产物14.7g,收率97%。1h nmr(400mhz,cdcl3):δ=1.43(s,18h),2.75
ꢀ–
2.91(m,8h),3.73(s,6h),6.03(s,2h),6.42(t,2h),6.77(s,2h)。
[0050]
实施例7:4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢茚-7,7
’‑
二酚(5)
[0051][0052]
在500ml的圆底瓶中,依次加入4(11.0g,22.2mmol),105g多聚磷酸,并将混合物在120℃下加热搅拌6小时。反应结束后,用水洗涤,乙酸乙酯萃取有机相,无水硫酸钠干燥,减压旋干得到粗品,并通过快速柱层析分离。用正己烷重结晶后得到目标产物22.8g,收率78%。1h nmr(400mhz,cdcl3):δ=1.37 (d,36h),2.31(m,4h),3.04
–
3.27(m,4h),5.44(s,2h),7.09(s,2h)。
[0053]
实施例8:4,4
’‑
二甲氧基-6,6
’‑
二叔丁基-1,1
’‑
螺二氢茚-7,7
’‑
二酚(5a)
[0054][0055]
中间体5a的合成步骤与中间体5相似,中间体4a投料11.0g,最终得到目标产物14.7g,收率81%。1h nmr(400mhz,cdcl3):δ=1.44(s,18h),2.52
–
2.18 (m,4h),2.91
–
3.11(m,4h),3.79(s,6h),5.25(s,2h),6.66(s,2h)。
[0056]
实施例9:7,7
’‑
二[(1,1
’‑
联苯-2,2
’‑
二基)亚膦酸酯]-4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢茚的制备(6-l4)
[0057][0058]
在一个干燥的500ml的schlenk瓶中于氮气保护下依次加入5(5.0g,10.5 mmol),无水三乙胺(21.9ml,157.5mmol,15eq.)和无水四氢呋喃80ml。然后,将混合物冷却至-40℃后滴加1,1
′‑
二氧基氯化膦(6.6g,26.3mmol,2.5equiv.) 的60ml无水四氢呋喃溶液中,滴完后室温反应24小时,氮气氛围下将反应液浓缩,粗品快速柱层析分离后,用乙腈重结晶得到目标产物5.6g,收率59%。1h nmr(600mhz,cdcl3):δ=1.42(d,36h),2.35(m,4h),3.18(m,4h),6.97
–ꢀ
7.10(m,8h),7.15(s,2h),7.30(td,4h),7.68(dd,4h);
31
p nmr(243mhz,cdcl3): δ=142.67。
[0059]
实施例10:7,7
’‑
二[(1,1
’‑
联苯-2,2
’‑
二基)亚膦酸酯]-4,4
’‑
二甲氧基-6,6
’‑
二叔丁基-1,1
’‑
螺二氢茚的制备(6a-l4)
[0060][0061]
配体6a-l4的合成步骤与中间体6-l4相似,螺二酚5a投料5.0g,最终得到目标产物4.6g,收率46%。1h nmr(600mhz,cdcl3):δ=1.45(s,18h),2.37(m, 4h),3.02(m,4h),3.78(s,6h),6.70(s,2h),6.97
–
7.13(m,8h),7.30(td,4h),7.69 (dd,4h);
31
p nmr(243mhz,cdcl3):δ=140.59。
[0062]
这里要指出的是,通式ii中其它的l1-l31的螺环双亚膦酸酯配体,只需要使用不
同的氯代亚膦酸酯取代基衍生物即可制备得到。
[0063]
在得到目标螺环双亚膦酸酯配体后,我们开发出了与这种新型螺环双亚膦酸酯配体相配套的间歇式小试反应装置(说明书附图),模拟工业上混合/醚后碳四的氢甲酰化反应。我们分别使用了两种碳四原料,第一种为醚后碳四,组分含量分别为(w/w):异丁烷(52.1%)、1-丁烯(16.6%)、顺-2-丁烯(15.3%)和反-2
‑ꢀ
丁烯(16.0%);第二种为mto碳四,组分含量分别为(w/w):正丁烷(6.0%)、 1-丁烯(0.7%)、顺-2-丁烯(34.7%)和反-2-丁烯(58.6%)。
[0064]
为保证配体活性以及醛产物不被氧化,以上物料通过原料预处理装置,除了需要除水、除氧,以及除硫(硫化物)、除氯(卤化物)、除含氮化合物(如: hcn)等以外,还需除去碳四原料中对铑催化剂有抑制作用的羧酸、丁二烯、丙二烯、炔烃等物质。为了测试新型螺环双亚膦酸酯配体在醚后/mto碳四中的反应活性,我们在接近相同的反应条件下对比测试其它商业化和专利文献报道过的配体,在以下实施例中所使用的配体ligand 1-10具有如下的结构:
[0065][0066]
本对比实施例使用上述实施例所列举的螺环双亚膦酸酯作为过渡金属配位体催化烯烃发生氢甲酰化反应,具体如下:
[0067]
对比实施例1:在氩气氛围下向200ml装有压力传感器、温度探针、在线取样口和安全泄压阀等装置的不锈钢高压反应釜中加入一定量的rh(acac)(co)2(0.01 mmol,2.6mg)和一定量的配体ligand 1-10(0.03
–
0.04mmol),加入一定体积的甲苯和内标物正癸烷,用磁子搅拌络合30分钟,生成铑与配体的催化络合物。随后,连接气体管线并充分置换后,在两位四通阀的切换下,用带计量功能的柱塞泵向反应釜内加入一定比例液态化的醚后碳四,使铑催化剂在总溶液中的浓度控制在159ppm左右,再室温均匀搅拌5~10分钟。搅拌均匀后,向反应装置内充入一氧化碳与氢气的混合气(1:1)至总压为1.0mpa。用磁力搅拌器
(加热釜底部)和电加热套(加热釜体)将反应釜升至所需温度(70℃),反应中持续补气保持总压力恒定在1.0mpa。反应2~4个小时后,将反应釜接入-40℃冷套降温,待釜温降至常温后,在不开釜的情况下,打开在线取样口取样,用色谱级的乙酸乙酯稀释后,气相色谱仪(gc)测定正异比(正戊醛/2-甲基丁醛的比例:l:b)。开釜后,在通风橱内将高压反应釜内的气体释放完全,取样称重。结果如表1所示。
[0068]
表1
[0069][0070][0071]
对比实施例2:在氩气氛围下向200ml装有压力传感器、温度探针、在线取样口和安全泄压阀等装置的不锈钢高压反应釜中加入一定量的rh(acac)(co)2(0.01 mmol,2.6mg)和一定量的配体ligand 1-10(0.03
–
0.04mmol),加入一定体积的甲苯和内标物正癸烷,用磁子搅拌络合30分钟,生成铑与配体的催化络合物。随后,连接气体管线并充分置换后,在两位四通阀的切换下,用带计量功能的柱塞泵向反应釜内加入一定比例液态化的mto碳四,使铑催化剂在总溶液中的浓度控制在159ppm左右,再室温均匀搅拌5~10分钟。搅拌均匀后,向反应装置内充入一氧化碳与氢气的混合气(1:1)至总压为1.0mpa。用磁力搅拌器(加热釜底部)和电加热套(加热釜体)将反应釜升至所需温度(70℃),反应中持续补气保持总压力恒定在1.0mpa。反应2~4个小时后,将反应釜接入-40℃冷套降温,待釜温降至常温后,在不开釜的情况下,打开在线取样口取样,用色谱级的乙酸乙酯稀释后,气相色谱仪(gc)测定正异比(正戊醛/2-甲基丁醛的比例:l: b)。开釜后,在通风橱内将高压反应釜内的气体释放完全,取样称重。结果如表 2所示。
[0072]
表2
[0073][0074]
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。