1.本发明涉及可用作药物的次黄嘌呤化合物的晶体。
2.更具体而言,本发明涉及具有脯氨酰羟化酶抑制作用并且可用作溃疡性结肠炎等炎症性肠病的治疗剂的次黄嘌呤化合物的晶体。
背景技术:
3.炎症性肠病(ibd)是由于过度免疫应答而导致肠黏膜发生炎症和溃疡的慢性疾病。ibd包括例如溃疡性结肠炎和克罗恩病。
4.溃疡性结肠炎是一种引起不明原因的弥漫性非特异性炎症的大肠疾病。大肠粘膜溃烂,可在粘膜中形成糜烂或溃疡。溃疡性结肠炎可分为观察到血便、糜烂、溃疡等的“活动期”和活动期所见消失的“缓解期”。因为在病程中经常反复出现复发和缓解,所以需要长期治疗。
5.对于溃疡性结肠炎的治疗,5-氨基水杨酸制剂(5-asa)被首先用作标准药。然而,据报道,5-asa的有效性约为46%至64%,而通过施用5-asa得到缓解的患者不超过29%至45%。在观察不到5-asa的效果时,使用类固醇。除了这些药剂外,免疫抑制剂、tnf-α抗体等有时也用于治疗溃疡性结肠炎。然而,所有这些药剂都存在诸如副作用和需要谨慎施用等的问题。因此,需要对溃疡性结肠炎具有新作用方式的治疗剂。
6.已知在ibd的病理状态下,低氧诱导因子1α(hif-1α)诱导与胃肠道上皮的屏障功能相关的基因表达。hif-1α是低氧诱导因子α(hif-α)的亚型之一。hif-α在低氧的环境(低氧)中稳定,然后它响应低氧而激活各种基因的转录。另一方面,hif-α的脯氨酸残基在富氧环境(常氧)中被脯氨酰羟化酶(phd)水解,然后通过蛋白酶体途径降解hif-α。
7.已知phd有三种亚型,即phd1、phd2和phd3。已知akb-4924为phd抑制剂。据报道,akb-4924具有phd2抑制作用,并稳定大肠组织中的hif-1α(非专利文献1)。此外,akb-4924对tnbs诱导的结肠炎模型具有改善效果。
8.另一方面,phd抑制剂例如罗沙司他(roxadustat)和达普司他(daprodustat)具有造血作用,已被开发为贫血治疗剂(非专利文献2)。因此,当phd抑制剂用作ibd治疗剂时,避免造血作用等的全身作用是重要的。
9.例如,作为phd抑制剂,专利文献1中描述了喹唑啉酮化合物,专利文献2中描述了吡唑并嘧啶化合物。专利文献3至7和非专利文献3中描述或例示了包含次黄嘌呤的化合物。然而,上述文献中未描述本发明的次黄嘌呤化合物的晶体。
10.现有技术文献
11.专利文献
12.专利文献1:国际公布第2010/093727号
13.专利文献2:美国公布的第2015/0239889号申请
14.专利文献3:美国公布的第2015/0368247号申请
15.专利文献4:美国公布的第2013/0165426号申请
16.专利文献5:美国公布的第2010/0029671号申请
17.专利文献6:美国公布的第2006/0258651号申请
18.专利文献7:美国公布的第2010/0120761号申请
19.非专利文献
20.非专利文献1:ellen marks等,"inflamm.bowel.dis."2015年,第21卷,第2号,267-275页
21.非专利文献2:mun chiang chan等,"molecular aspects of medicine"2016年,第47-48卷,54-75页
22.非专利文献3:takashi goi等,"synlett",2018年,第29卷,第14号,1867-1870页
技术实现要素:
23.发明要解决的问题
24.本技术人开发了下式(i)所示的化合物(以下称为“化合物1”)作为具有phd2抑制作用的新型化合物,并提交了发明专利申请(pct/jp2019/035995)。
25.[化学式1]
[0026][0027]
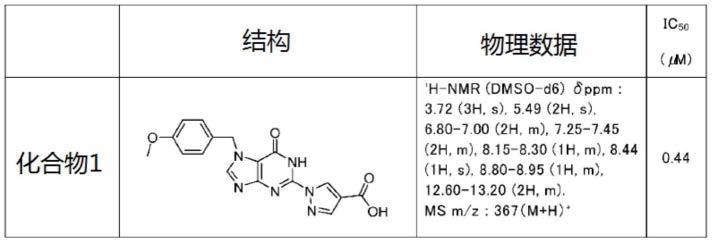
化学名:1-(7-(4-甲氧基苄基)-6-氧代-6,7-二氢-1h-嘌呤-2-基)-1h-吡唑-4-羧酸。
[0028]
作为原料药,具有良好物理性质的晶体是优选的。然而,作为原料药的最优异的晶形可因化合物而异。一般而言,预测具有良好物理性质的原料药的晶形是困难的,需要对各化合物进行种种检查。因此,本发明的目的在于提供具有良好物理性质的晶体作为新型化合物(化合物1)的原料药。
[0029]
解决问题的手段
[0030]
本发明涉及具有phd2抑制作用并且可用于治疗炎症性肠病的化合物1的晶体。亦即,本发明涉及以下[1]至[7]项等。
[0031]
[1]由下式(i)表示的化合物的晶体:
[0032]
[化学式2]
[0033][0034]
[2]上述[1]所述的晶体,其是在粉末x射线衍射中具有在选自以下(i)至(iv)的衍射角(2θ(
°
))处的峰的子集的所述化合物的晶体:
[0035]
(i)6.5
±
0.2和13.5
±
0.2的峰(晶形i);
[0036]
(ii)5.1
±
0.2和16.9
±
0.2的峰(晶形ii);
[0037]
(iii)5.8
±
0.2和17.7
±
0.2的峰(晶形iii);以及
[0038]
(iv)5.0
±
0.2和5.6
±
0.2的峰(晶形iv)。
[0039]
[3]上述[1]所述的晶体,其是在粉末x射线衍射中具有在选自以下(i)至(iv)的衍射角(2θ(
°
))处的峰的子集的所述化合物的晶体:
[0040]
(i)6.5
±
0.2和16.2
±
0.2的峰(晶形i);
[0041]
(ii)5.1
±
0.2和10.8
±
0.2的峰(晶形ii);
[0042]
(iii)5.8
±
0.2和25.4
±
0.2的峰(晶形iii);以及
[0043]
(iv)5.0
±
0.2、15.2
±
0.2和26.4
±
0.2的峰(晶形iv)。
[0044]
[4]上述[1]所述的晶体,其是在粉末x射线衍射中具有在5.8
±
0.2、8.4
±
0.2、13.6
±
0.2、17.0
±
0.2、17.4
±
0.2、17.7
±
0.2、25.4
±
0.2和25.8
±
0.2的衍射角(2θ(
°
))处的峰的所述化合物的晶体。
[0045]
[5]药物组合物,其包含上述[1]至[4]中任一项所述的晶体和药物添加剂。
[0046]
[6]上述[5]所述的药物组合物,其是用于治疗炎症性肠病的药物组合物。
[0047]
[7]上述[6]所述的药物组合物,其中所述炎症性肠病是溃疡性结肠炎或克罗恩病。
[0048]
在一个实施方式中,本发明涉及炎症性肠病的治疗方法,所述方法包括将必要量的上述[5]所述的药物组合物施用于患者。
[0049]
在一个实施方式中,本发明涉及上述[1]至[4]中任一项所述的化合物的晶体在制备用于治疗炎症性肠病的药物组合物中的用途。
[0050]
在一个实施方式中,本发明是上述[1]所述的晶体,其是在粉末x射线衍射中具有在选自以下(i)至(iv)的衍射角(2θ(
°
))处的峰的子集的所述化合物的晶体:
[0051]
(i)6.5
±
0.2、13.5
±
0.2、16.2
±
0.2和17.5
±
0.2的峰(晶形i);
[0052]
(ii)5.1
±
0.2、10.8
±
0.2、16.4
±
0.2和16.9
±
0.2的峰(晶形ii);
[0053]
(iii)5.8
±
0.2、13.6
±
0.2、17.7
±
0.2和25.4
±
0.2的峰(晶形iii);以及
[0054]
(iv)5.0
±
0.2、5.6
±
0.2、15.2
±
0.2和26.4
±
0.2的峰(晶形iv)。
[0055]
在一个实施方式中,本发明是上述[1]所述的晶体,其是在粉末x射线衍射中具有在衍射角(2θ(
°
))处的峰的子集并且吸热的所述化合物的晶体,其中所述峰的子集和所述吸热选自以下(i)至(iv):
[0056]
(i)6.5
±
0.2、13.5
±
0.2、16.2
±
0.2和17.5
±
0.2的峰以及300℃左右的吸热峰顶(晶形i);
[0057]
(ii)5.1
±
0.2、10.8
±
0.2、16.4
±
0.2和16.9
±
0.2的峰以及301℃左右的吸热峰顶(晶形ii);
[0058]
(iii)5.8
±
0.2、13.6
±
0.2、17.7
±
0.2和25.4
±
0.2的峰以及294℃左右的吸热峰顶(晶形iii);以及
[0059]
(iv)5.0
±
0.2、5.6
±
0.2、15.2
±
0.2和26.4
±
0.2的峰以及301℃左右的吸热峰顶(晶形iv);
[0060]
发明的效果
[0061]
本发明的化合物的晶体具有作为原料药的良好物理性质。
附图说明
[0062]
图1至图4是晶体的粉末x射线衍射图。纵轴表示衍射强度(计数)。横轴表示衍射角(2θ(
°
))。
[0063]
图5至图8是晶体的热重差热分析图(tg-dta测量图)。纵轴(左)表示热重(tg)曲线中的质量变化(%)。纵轴(右)表示差热分析(dta)曲线中的热流量(μv)。横轴表示温度(℃)。
[0064]
[图1]化合物1的晶形i的粉末x射线衍射图。
[0065]
[图2]化合物1的晶形ii的粉末x射线衍射图。
[0066]
[图3]化合物1的晶形iii的粉末x射线衍射图。
[0067]
[图4]化合物1的晶形iv的粉末x射线衍射图。
[0068]
[图5]化合物1的晶形i的tg-dta测量图。
[0069]
[图6]化合物1的晶形ii的tg-dta测量图。
[0070]
[图7]化合物1的晶形iii的tg-dta测量图。
[0071]
[图8]化合物1的晶形iv的tg-dta测量图。
具体实施方式
[0072]
以下更详细地描述本发明的实施方式。
[0073]
在本发明中,除非另有规定,否则各术语具有以下含义。
[0074]
在说明书、附图和表格中的以下缩写分别具有以下含义。
[0075]
cdcl3:氘代氯仿
[0076]
dmso:二甲基亚砜
[0077]
nmp:1-甲基-2-吡咯烷酮
[0078]
thf:四氢呋喃
[0079]
tnbs:三硝基苯磺酸
[0080]
pd2(dba)3:三(二亚苄基丙酮)二钯(0)
[0081]
tbuxphos:2-二叔丁基膦基-2’,4’,6
’‑
三异丙基联苯
[0082]
氨基硅胶:氨丙基化硅胶
[0083]
ods柱色谱:十八烷基甲硅烷基化硅胶柱色谱
[0084]
结构:结构式
[0085]
物理数据:物理数据
[0086]
ic
50
:50%抑制所需的浓度
[0087]
fitc:荧光素异硫氰酸酯
[0088]1h-nmr:氢核磁共振谱
[0089]
dmso-d6:二甲基亚砜-d6
[0090]
ms:质谱
[0091]
esi_apci:使用电喷雾电离-大气压化学电离的多电离
[0092]
在本发明中,“作为原料药,物理性质良好”是指,例如,晶体在试验例4所示的固体
稳定性试验中是物理化学稳定的或化学稳定的。
[0093]
本发明的化合物1的晶体还包括其与水或乙醇等药学上可接受的溶剂的溶剂合物。
[0094]
在本发明的化合物1中,一部分的原子可被相应的同位素置换。本发明包括其中的原子被这些同位素置换的化合物。同位素的例子包括由2h、3h、
11
c、
13
c、
14
c、
36
cl、
18
f、
123
i、
125
i、
13
n、
15
n、
15
o、
17
o、
18
o、
32
p和
35
s表示的氢原子、碳原子、氯原子、氟原子、碘原子、氮原子、氧原子、磷原子和硫原子的同位素。在一个实施方式中,可以举出化合物1的一部分氢原子被2h(d:氘原子)置换的化合物。
[0095]
一部分原子被同位素置换的本发明化合物1可以通过使用市售的导入同位素的结构单元进行与后述的制造方法相似的方法来制备。
[0096]
本发明的化合物1具有优异的phd2抑制作用,因此可以用作ibd的治疗剂(参见nature reviews drug discovery,2014,13,852-869页)。在本发明中,词语“ibd”包括例如溃疡性结肠炎、克罗恩病、肠白塞病、感染性肠炎、放射性肠炎、药物诱导的肠炎、缺血性肠炎、肠系膜静脉硬化(静脉硬化性结肠炎)、梗阻性结肠炎和胶原病引起的肠炎。优选地,本发明的化合物1可以用作溃疡性结肠炎或克罗恩病的治疗剂(参照inflamm.bowel.dis.,2015,21(2),267-275页)。
[0097]
在本发明中,词语“治疗”包括“预防”的含义。溃疡性结肠炎的治疗包括例如“预防复发”和“维持缓解”的含义。
[0098]
本发明的化合物1对结肠炎的治疗效果可以根据试验例2中描述的方法或本技术领域公知的方法来确定。例如,也可以根据biol.pharm.bull.,2004,27(10),1599-1603页等描述的方法或与其类似的方法来确定效果。
[0099]
在一个实施方式中,本发明的化合物1是phd2抑制剂,其特异性作用于大肠组织以限制hif-α稳定化的脱靶效应。术语“特异性作用于大肠组织”是指例如化合物在大肠组织中的浓度比在血中的浓度高,并且化合物对大肠发挥治疗效果但没有全身性作用(例如,造血作用)(参照试验例2和3)。
[0100]
取决于用法,本发明的药物组合物以各种剂型使用。作为这样的剂型,例如,可以举出粉剂、颗粒剂、细颗粒剂、干糖浆剂、片剂、胶囊剂、注射剂、液体剂、软膏剂、栓剂、贴敷剂和灌肠剂。优选地,本发明的药物组合物是口服施用的。
[0101]
本发明的药物组合物包含化合物1的晶体作为有效成分。
[0102]
本发明的药物组合物使用化合物1的晶体和至少一种药物添加剂制备。取决于它们的剂型,这些药物组合物可以根据公知的制剂程序,使用适当的药物添加剂如赋形剂、崩解剂、粘合剂、润滑剂、稀释剂、缓冲剂、张度剂、防腐剂、润湿剂、乳化剂、分散剂、稳定剂、增溶剂等适当地混合、稀释或溶解来配制。
[0103]
当本发明的药物组合物用于治疗时,化合物1的剂量取决于各患者的年龄、性别、体重、以及疾患和治疗的程度等来适当地决定。日剂量可以分成每天一次、两次、三次、四次来施用。
[0104]
在口服施用的情况下,成人的剂量可以确定在例如每天0.1至1000mg范围内。在一个实施方式中,口服施用剂量可以确定在每天1至500mg范围内,优选在每天10至200mg范围内。
[0105]
在胃肠外施用的情况下,成人的剂量可以确定为例如每天0.1至1000mg。在一个实施方式中,胃肠外施用剂量可以确定在每天0.5至200mg范围内,优选在每天1至20mg范围内。
[0106]
在一个实施方式中,本发明的药物组合物还可以与除phd抑制剂以外的任何其它药物组合使用。作为可以组合用于治疗炎症性肠病的其它药剂,可以举出例如5-asa、类固醇、免疫抑制剂、tnf-α抗体、janus激酶抑制剂和α4β7整联蛋白抗体。
[0107]
当本发明的化合物1的晶体与其它药剂组合使用时,它们可以作为含有这些活性成分的一体化制剂或作为各自从每种活性成分分别配制的制剂施用。当分别配制时,这些制剂可以分开或同时施用。此外,本发明的化合物1的剂量可以取决于组合使用的其它药剂的剂量而适当减量。
[0108]
粉末x射线分析测量
[0109]
对于粉末x射线衍射,将晶体用研钵磨碎,然后用粉末x射线衍射装置smartlab(rigaku)通过反射法并根据以下条件测量。
[0110]
x射线源,波长:cukα射线(cukα1和cukα2),
[0111]
管电压、管电流、扫描速度:40kv,50ma,13
°
2(θ)/min
[0112]
数据分析软件:smartlab studio ii(rigaku)
[0113]
数据分析方法(峰定义):峰位置(峰顶位置、用cukα1和cukα2辐照时的衍射角)、峰高(不含背景)
[0114]
众所周知,粉末x射线衍射图样中的峰的相对强度(相对峰高)可因样品条件和测量条件而变动。相对强度会因晶体生长方向、粒子大小、测量条件等而略有变化,因此不应被严格地解读。
[0115]
粉末x射线衍射中衍射图样的2θ值可能因样品条件和测量条件而略有变动,是公知的。一般而言,2θ值可在约
±
0.2(
°
)的范围内变动。因此,本发明不仅包括在粉末x射线衍射中峰的衍射角(2θ(
°
))完全重合的晶体,而且包括全部或一部分峰的衍射角(2θ(
°
))在
±
0.2(
°
)的范围内重合的晶体。
[0116]
热分析测量(热重差热分析(tg-dta))
[0117]
在氮气氛下使用差热天平(tg8120型或thermo plus evo2型,rigaku),根据以下测量条件进行热分析。
[0118]
升温速度:10℃/min
[0119]
基准物质:氧化铝
[0120]
测量质量减少开始时的温度和其会聚于一点时的温度,并由所述温度下的质量差来计算质量变化(%)。
[0121]
在tg-dta测量图中,dta曲线中的“吸热”由峰顶点处温度(峰顶)或“外推起始温度”表示。“外推起始温度”是指dta曲线中的起始点或偏移点与基线外推之间的交点,也称为“外推开始温度”。“外推起始温度”是峰的起始点的温度,是指通过外推算出的放热或吸热起始温度。tg-dta测量图中的峰顶和外推起始温度可能会因测量条件而略有波动。例如,一般而言,所述温度可在
±
5℃的范围内波动。因此,由上述峰指定的晶体包括在
±
5℃的范围内重合的晶体。
[0122]
在本发明中,所述热分析中使用的“左右”是指
±
5℃的范围,优选是指
±
2℃。
[0123]
本发明的化合物1的晶体的晶形可以由各实施例中描述的粉末x射线衍射中的衍射峰的子集来指定。在本发明中,“具有峰的子集”是指晶体至少包含该子集的峰作为特征峰。
[0124]
此外,还可以通过在粉末x射线衍射中的衍射峰的子集和各晶体的tg-dta测量中的吸热等物理性质的组合,来指定晶形。
[0125]
实施例
[0126]
通过以下实施例进一步更详细地说明本发明。然而,本发明不限于此。
[0127]
参考例1
[0128]
2,6-二氯-7-(4-甲氧基苄基)-7h-嘌呤
[0129]
在室温下向4-甲氧基苄基氯(9.40g)在thf(100ml)中的溶液添加碘化钠(9.74g)。将反应混合物搅拌1小时后,添加2,6-二氯嘌呤(9.45g)和碳酸钾(10.37g)。将反应混合物在室温搅拌20小时。向反应混合物添加水并用乙酸乙酯提取。将有机层用饱和盐水洗涤,经无水硫酸镁干燥,然后减压浓缩。所得残余物通过硅胶柱色谱法纯化(洗脱液:乙酸乙酯/己烷=37/63-58/42-70/30),各得到2,6-二氯-9-(4-甲氧基苄基)-9h-嘌呤(2.48g)和标题化合物(2.22g)。
[0130]1h-nmr(cdcl3)δppm:3.82(3h,s),5.59(2h,s),6.80-7.00(2h,m),7.05-7.25(2h,m),8.18(1h,s)。
[0131]
参考例2
[0132]
2-氯-6-甲氧基-7-(4-甲氧基苄基)-7h-嘌呤
[0133]
在氩气气氛和冰冷却下,向参考例1(4.35g)在thf(70ml)中的溶液滴加28%的甲醇钠甲醇溶液(2.58g)。将反应混合物在室温搅拌30分钟。向反应混合物添加水,通过过滤收集不溶物。将所得的固体用水洗涤,然后在50℃减压干燥6小时,得到标题化合物(3.47g)。
[0134]
参考例3
[0135]
1-(6-甲氧基-7-(4-甲氧基苄基)-7h-嘌呤-2-基)-1h-吡唑-4-甲酸乙酯
[0136]
在氩气气氛下,向参考例2(3.21g)、1h-吡唑-4-甲酸乙酯(1.77g)和tbuxphos(0.90g)在nmp(21ml)中的溶液添加磷酸三钾(3.35g)和pd2(dba)3(0.48g)。将反应混合物在60℃搅拌3小时。使反应混合物冷却至室温后,向反应混合物添加水和乙酸乙酯,通过hyflo super-cel滤去不溶物。滤液用乙酸乙酯提取后,用水和盐水洗涤有机层。将有机层通过氨基硅胶,并减压浓缩。向所得的残余物添加乙醇,通过过滤收集不溶物。将所得的固体用乙醇和二异丙醚洗涤,然后通过氨基硅胶柱色谱法纯化(洗脱液:乙酸乙酯/己烷=81/19-100/0),得到标题化合物(2.02g)。
[0137]
化合物1
[0138]
1-(7-(4-甲氧基苄基)-6-氧代-6,7-二氢-1h-嘌呤-2-基)-1h-吡唑-4-羧酸
[0139]
向参考例3(4.48g)在thf(55ml)中的溶液添加水(27ml)和氢氧化锂水溶液(4mol/l,27ml)。将反应混合物在50℃搅拌18小时。使反应混合物冷却至室温后,向反应混合物添加2mol/l盐酸(66ml)并在室温下搅拌1小时。通过过滤收集所生成的不溶物。将所得的固体用水洗涤,然后在50℃减压干燥8小时,得到标题化合物(3.87g)。
[0140]
[表1]
[0141][0142]
试验例1:phd2抑制试验
[0143]
(1)人phd2
184-418
的表达和制备
[0144]
通过以下方法来表达和制备含有由cac42509(genbank登录id)表示的蛋白质的氨基酸残基184至418的人phd2
184-418
。
[0145]
将含有n-末端组氨酸标签的人phd2
184-418
的表达构建体引入pet-30a( )载体,并确认序列。将该载体引入bl21(de3)菌株并在含有抗生素的lb培养基中37℃培养。培养后,向细胞添加细胞裂解液,然后通过超声波使细胞破碎和悬浮。将破碎的悬浮液离心,上清液通过ni柱纯化,得到人phd2
184-418
。
[0146]
(2)试验方法
[0147]
人hif-1α
556-574
(fitc-标记的hif-1α
556-574
)含有人hif-1α的氨基酸残基556至574(部分肽)并且在该hif-1α
556-574
的n-末端含有fitc-ahx,将其用作底物。使用fitc-标记的hif-1α
556-574
,基于fitc-标记的hif-1α
556-574
的荧光偏振变化,通过以下方法评价2-酮戊二酸与试验化合物(phd抑制剂)之间的竞争性抑制。
[0148]
将酶(人phd2
184-418
)和所述底物用含有10mm hepes、150mm nacl、10μm mncl
2-4h2o、2μm 2-酮戊二酸和0.05%tween-20的测定缓冲液(ph 7.4)进行稀释。试验化合物用dmso稀释。将试验化合物和人phd2
184-418
预先添加到384孔板(corning,黑色,不透明底)。通过添加fitc-标记的hif-1α
556-574
开始反应。在37℃温育60分钟后,通过pherastar fsx(bmg labtech)测量荧光偏振(激发波长:470nm,荧光波长:530nm)。测量各孔的荧光偏振,并基于无试验物质组的值来计算试验化合物的人phd2结合抑制活性。
[0149]
(3)结果
[0150]
如表1所示,本发明的化合物1抑制phd2与hif-1α之间的结合。因此,证明本发明的化合物1可用作phd2抑制剂。
[0151]
试验例2:在结肠炎模型中的治疗效果
[0152]
(1)tnbs诱导的结肠炎模型大鼠
[0153]
已知当将tnbs施用于大鼠的大肠内时,在大肠局部引起炎症,然后由于肠道内屏障功能的破坏,肠粘膜透过性增加。因此,评价口服施用试验化合物对肠粘膜透过性的抑制作用作为药效的指标。
[0154]
(2)试验方法
[0155]
使用sd雄性大鼠:8周龄的slc(日本slc)。在戊巴比妥麻醉下,在大肠内距肛门8cm的部位施用300μl用50%乙醇制备的28mg/ml tnbs,以引起炎症。对溶剂处理组施用300μl的50%乙醇。将动物在施用tnbs之前禁食48小时。从次日起,每天一次口服施用以0.05%甲基纤维素溶液制备的试验化合物(3mg/kg),共施用3天。施用3天后,在施用试验化合物后4
小时,口服施用50mg/kg fitc。4小时后在异氟醚麻醉下从颈静脉采集血样。离心分离血清,通过pherastar fsx(bmg labtech)检测荧光强度以测量透过肠系膜渗入循环血液中的fitc浓度。试验化合物对肠粘膜透过性的抑制率以无试验物质组的值为0并以tnbs未处理组的值为100进行计算。
[0156]
(3)结果
[0157]
试验化合物对肠粘膜透过性的抑制率(%平均值)(抑制)如下所示。
[0158]
[表2]
[0159] 抑制(%)化合物183
[0160]
施用本发明的化合物1,抑制了因施用tnbs而增加的fitc的肠粘膜透过性。因此,证明本发明的化合物1可用作炎症性肠病的治疗剂。
[0161]
试验例3:大肠组织中的化合物浓度
[0162]
(1)大鼠pk试验
[0163]
将用0.05%甲基纤维素制备的试验化合物(3mg/kg/5ml)口服施用于未禁食的大鼠(sd,8周龄,雄性,日本slc)。施用后0.25、0.5、1、2、4、6和8小时从颈静脉采集血样。在异氟醚麻醉下行开腹术,分离大肠。将收集的远端大肠(约5cm)切开,然后将提取的大肠在盘子上用生理盐水洗涤。洗涤后,用小剪刀将大肠剪碎。将其中约150mg移至试管。向该试管添加100μl生理盐水,并使用摇匀器(1000rpm
×
30分钟)将该混合物均质化。通过添加四倍体积的生理盐水作为最终体积来制备样本。通过使用液相色谱-质谱(lc/ms)的定量分析来测量大肠组织和血浆中的试验化合物浓度。
[0164]
作为比较例,使用us2015/0239889(wo2014/030716)中作为实施例55描述的化合物(化合物a)。
[0165]
(2)大肠组织和血浆中的化合物浓度
[0166]
如下表所示,证明本发明的化合物1在大肠组织中的浓度高于在血浆中的浓度。因此,本发明的化合物1是特异性作用于大肠组织的phd2抑制剂。
[0167]
[表3]
[0168] cmaxaucplasmacolonc/p化合物1101.095<1209>209化合物a28179.95574<50<0.7
[0169]
表中符号具有以下含义。
[0170]
化合物a:比较例
[0171]
cmax:试验化合物在口服施用的情况下的最大血浆浓度(ng/ml)
[0172]
auc:血浆试验化合物浓度-时间曲线下面积(ng*min/ml)
[0173]
plasma:8小时后血浆中试验化合物浓度(ng/ml)
[0174]
colon:8小时后大肠组织中试验化合物浓度(ng/g)
[0175]
c/p:上述colon与plasma之比
[0176]
实施例1-1:化合物1的晶形i
[0177]
将参考例3(4.90g)、thf(40ml)、氢氧化锂水溶液(4mol/l,30ml)和水(15ml)的混合物在60℃搅拌4小时。将反应混合物加热至50℃,分小份滴加2mol/l盐酸以达到ph 2。当
滴加2mol/l盐酸(38ml)时,将反应混合物通过硅藻土过滤。将硅藻土用水(20ml)洗涤,并将滤液与之前的滤液合并。向所得的滤液滴加2mol/l盐酸(包括上述38ml在内共72ml)。将所得的悬浮液在50℃搅拌1小时并在室温搅拌12小时后,对该浆液进行抽滤。将所得的固体用水(10ml)洗涤两次,然后用水(30ml)洗涤3次。将所得的固体在50℃减压干燥,得到化合物1的晶形i(4.33g)。
[0178]
测量化合物1的晶形i的粉末x射线衍射。主要衍射峰的衍射角(2θ(
°
))和该衍射峰的相对强度(rel.den.(%))如表4所示。
[0179]
[表4]
[0180][0181]
对于化合物1的晶形i的鉴定,例如,可以使用选自以下[1-1-1]至[1-1-6]的衍射峰子集:
[0182]
[1-1-1]6.5
±
0.2和13.5
±
0.2的峰;
[0183]
[1-1-2]6.5
±
0.2和16.2
±
0.2的峰;
[0184]
[1-1-3]6.5
±
0.2、13.5
±
0.2和16.2
±
0.2的峰;
[0185]
[1-1-4]6.5
±
0.2、13.5
±
0.2、16.2
±
0.2和17.5
±
0.2的峰;
[0186]
[1-1-5]6.5
±
0.2、9.7
±
0.2、13.5
±
0.2、16.2
±
0.2和17.5
±
0.2的峰;以及
[0187]
[1-1-6]6.5
±
0.2、9.7
±
0.2、13.5
±
0.2、16.2
±
0.2、17.5
±
0.2和19.9
±
0.2的峰。
[0188]
进行化合物1的晶形i的热分析。
[0189]
吸热:300℃左右(峰顶,外推起始温度298℃左右)
[0190]
质量减少:30℃至280℃左右(0.7%)
[0191]
实施例1-2:化合物1的晶形ii
[0192]
将化合物1(10mg)、1,4-二烷(0.9ml)、水(0.1ml)和2mol/l氢氧化钠水溶液(27.5μl)的悬浮液在60℃搅拌。溶解后,将溶液过滤。在40℃向滤液添加2mol/l盐酸(41μl)。将混合物在相同温度下搅拌2小时,然后在室温下搅拌60小时。对该浆液进行抽滤,得到化合物1的晶形ii。将所得的晶形ii用作下一步操作的种晶。
[0193]
将化合物1(150mg)、1,4-二烷(3ml)、水(1.5ml)和2mol/l氢氧化钠水溶液(413μl)的悬浮液在60℃搅拌。溶解后,将溶液过滤。在40℃向滤液添加2mol/l盐酸(413μl)。在相同温度下,添加种晶(1mg),并将溶液搅拌1小时。在室温下向该混合物添加2mol/l盐酸(207μl),并搅拌混合物2小时。对浆液进行抽滤,所得的固体用水(5ml)洗涤两次。将所得的固体在50℃减压干燥,得到化合物1的晶型ii(118mg)。
[0194]
测量化合物1的晶形ii的粉末x射线衍射。主要衍射峰的衍射角(2θ(
°
))和该衍射
峰的相对强度(rel.den.(%))如表5所示。
[0195]
[表5]
[0196][0197]
对于化合物1的晶形ii的鉴定,例如,可以使用选自以下[1-2-1]至[1-2-6]的衍射峰子集:
[0198]
[1-2-1]5.1
±
0.2和16.9
±
0.2的峰;
[0199]
[1-2-2]5.1
±
0.2和10.8
±
0.2的峰;
[0200]
[1-2-3]5.1
±
0.2、16.4
±
0.2和16.9
±
0.2的峰;
[0201]
[1-2-4]5.1
±
0.2、10.8
±
0.2、16.4
±
0.2和16.9
±
0.2的峰;
[0202]
[1-2-5]5.1
±
0.2、8.6
±
0.2、10.8
±
0.2、16.4
±
0.2和16.9
±
0.2的峰;以及
[0203]
[1-2-6]5.1
±
0.2、8.6
±
0.2、10.8
±
0.2、16.4
±
0.2、16.9
±
0.2和26.7
±
0.2的峰。
[0204]
进行化合物1的晶形ii的热分析。
[0205]
吸热:301℃左右(峰顶,外推起始温度300℃左右)
[0206]
质量减少:30℃至290℃左右(0.6%)
[0207]
实施例1-3:化合物1的晶形iii
[0208]
将化合物1(20mg)、甲醇(0.4ml)、水(0.2ml)和三乙胺(15μl)的悬浮液在60℃搅拌。溶解后,向溶液添加乙酸(10μl),并将该溶液在相同温度下搅拌5天。收集该混合物中产生的晶体,得到化合物1的晶形iii。将所得的晶型iii用作下一步操作的种晶。
[0209]
将化合物1的晶形i(0.2g)、甲醇(8ml)和晶形iii的种晶(1mg)的混合物在75℃搅拌约2小时。将混合物在65℃搅拌,向混合物添加种晶(1mg),并搅拌该混合物2天。将该悬浮液冷却至室温并抽滤。将所得的固体用甲醇(2ml)洗涤,在50℃减压干燥,得到化合物1的晶形iii(157mg)。
[0210]
测量化合物1的晶形iii的粉末x射线衍射。主要衍射峰的衍射角(2θ(
°
))和该衍射峰的相对强度(rel.den.(%))如表6所示。
[0211]
[表6]
[0212]
2θ(
°
)相对强度(%)2θ(
°
)相对强度(%)5.810017.4308.42517.73113.63225.43717.02325.822
[0213]
对于化合物1的晶形iii的鉴定,例如,可以使用选自以下[1-3-1]至[1-3-6]的衍
射峰子集:
[0214]
[1-3-1]5.8
±
0.2和17.7
±
0.2的峰;
[0215]
[1-3-2]5.8
±
0.2和25.4
±
0.2的峰;
[0216]
[1-3-3]5.8
±
0.2、17.7
±
0.2和25.4
±
0.2的峰;
[0217]
[1-3-4]5.8
±
0.2、13.6
±
0.2、17.7
±
0.2和25.4
±
0.2的峰;
[0218]
[1-3-5]5.8
±
0.2、8.4
±
0.2、13.6
±
0.2、17.7
±
0.2和25.4
±
0.2的峰;以及
[0219]
[1-3-6]5.8
±
0.2、8.4
±
0.2、13.6
±
0.2、17.0
±
0.2、17.4
±
0.2、17.7
±
0.2、25.4
±
0.2和25.8
±
0.2的峰。
[0220]
进行化合物1的晶形iii的热分析。
[0221]
吸热:294℃左右(峰顶,外推起始温度291℃左右)
[0222]
质量减少:30℃至280℃左右(0.2%)
[0223]
实施例1-4:化合物1的晶形iv
[0224]
将化合物1(600mg)悬浮在3ml甲醇中。在室温下向该悬浮液添加1mol/l氢氧化钠水溶液(3.28ml),然后添加水(0.9ml)。溶解后,在室温下向溶液添加1mol/l盐酸(1.64ml)。立即观察到沉淀。将该混合物在50℃搅拌1小时,向混合物添加甲醇(3ml)。将混合物在50℃再搅拌1小时,并向混合物添加甲醇(0.6ml)。将混合物在室温搅拌1小时。对浆液进行抽滤。所得的固体用30%含水甲醇洗涤,40℃减压干燥2小时,得到化合物1的钠盐的晶形i(471mg)。
[0225]
将该化合物1的钠盐的晶形i(59mg)悬浮于日本药典溶出试验的第1液(6ml)中,将该悬浮液37℃振荡搅拌15小时。过滤收集固体,将所得的固体在40℃减压干燥2小时,得到化合物1的晶形iv(47mg)。
[0226]
测量化合物1的甲磺酸盐的晶形i的粉末x射线衍射。主要衍射峰的衍射角(2θ(
°
))和该衍射峰的相对强度(rel.den.(%))如表7所示。
[0227]
[表7]
[0228]
2θ(
°
)相对强度(%)2θ(
°
)相对强度(%)5.010016.3285.66519.62814.02420.63515.23226.473
[0229]
对于化合物1的晶形iv的鉴定,例如,可以使用选自以下[1-4-1]至[1-4-5]的衍射峰子集:
[0230]
[1-4-1]5.0
±
0.2和5.6
±
0.2的峰;
[0231]
[1-4-2]5.0
±
0.2、15.2
±
0.2和26.4
±
0.2的峰;
[0232]
[1-4-3]5.0
±
0.2、5.6
±
0.2、15.2
±
0.2和26.4
±
0.2的峰;
[0233]
[1-4-4]5.0
±
0.2、5.6
±
0.2、15.2
±
0.2、16.3
±
0.2、20.6
±
0.2和26.4
±
0.2的峰;以及
[0234]
[1-4-5]5.0
±
0.2、5.6
±
0.2、14.0
±
0.2、15.2
±
0.2、16.3
±
0.2、19.6
±
0.2、20.6
±
0.2和26.4
±
0.2的峰。
[0235]
进行化合物1的晶形iv的热分析。
[0236]
吸热:235℃左右(峰顶),301℃左右(峰顶,外推起始温度299℃左右)
[0237]
质量减少:25℃至100℃左右(0.5%)
[0238]
试验例4:固体稳定性试验
[0239]
对化合物1的晶体(晶形i至iv)的物理化学稳定性和化学稳定性进行了调查。
[0240]
(1)方法
[0241]
物理化学稳定性:各晶体在40℃的开放条件下保存。在开始时和1个月后测量样品的粉末x射线衍射,并确认晶形变化。同时也观察性状的变化。
[0242]
化学稳定性:各晶体在40℃的开放条件下保存。在开始时和1个月后,在以下hplc测量条件下测量化合物1的质量。
[0243]
[hplc条件]
[0244]
检测器:紫外可见光吸光光度计/波长:225nm
[0245]
柱子:l-column2 ods,3μm,4.6
×
150mm(化学物质评价研究机构)
[0246]
柱温:恒温40℃左右
[0247]
流量:1.0ml/min
[0248]
流动相a:磷酸二氢钾溶解在水中至20mmol/l后,用磷酸制备ph 2.3的溶液
[0249]
流动相b:乙腈
[0250]
流动相比率:
[0251]
0至30分钟:流动相a/流动相b=81/19
[0252]
30至50分钟:流动相a/流动相b=81/19变为25/75
[0253]
50至55分钟:流动相a/流动相b=25/75
[0254]
55至55.01分钟:流动相a/流动相b=25/75变为81/19
[0255]
55.01至75分钟:流动相a/流动相b=81/19
[0256]
进样量:5μl
[0257]
样品冷却器:4℃
[0258]
溶解溶剂:混合溶液(流动相a/乙腈=75/25)
[0259]
样品溶液:样品溶解在溶解溶剂中并调节至约1.0mg/ml
[0260]
排除来自于空白的峰,通过自动积分来测量化合物1和类似物质的峰面积,并通过面积归一法计算化合物1的质量。另外,计算保存前后的化合物1的质量变化量。
[0261]
(2)结果
[0262]
在40℃的开放条件下保存,化合物1的晶体物理化学稳定,晶形和性状几乎没有变化。另外,化合物1的晶体化学稳定,化合物1的量几乎没有减少(表8)。
[0263]
因此,证明了本发明的化合物1的晶体具有作为原料药的良好物理性质。
[0264]
[表8]
[0265][0266]
产业可利用性
[0267]
本发明的化合物1的晶体具有作为原料药的良好物理性质,可用作炎症性肠病的治疗剂。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
