1.本发明涉及药物信息技术领域,具体涉及一种药物吸收速率常数的直接估测方法。
背景技术:
2.药物吸收是指药物自给药部位摄取并进入血液循环的过程。药物的吸收速率是药代动力学和生物利用度研究的一个重要参数。吸收速率对血药浓度影响很大,它受给药途径和许多其他因素的影响。给药途径不同,吸收速率自然不同;剂型不同,吸收速率亦各异。如固体剂型的口服吸收,就取决于制剂的崩解、活性药物的溶出、在吸收部位的药物浓度和血液循环,以及吸收面的位置和面积等等。若药物不吸收,当然不能起全身作用;如果吸收差,则必须加大剂量;要是吸收慢,可能作用会延迟;吸收过快,可能发生副作用;吸收不规则,又可能无法预测临床效应。因此,了解药物吸收速率,是临床前和临床研究的一个重要组成部分。
3.药物吸收速率常数ka值被描述为药物在血管外给药(即口服、经口、直肠等)后通过黏膜进入体循环的速率,最终影响达峰时间(t
max
)和达峰浓度体内药物(c
max
)。药物吸收率常数(ka)的定量评估在制药工业中起着至关重要的作用。例如,使用一种剂型的体内吸收率和体外溶出率(ivivc)之间的相关性来预测药物的生物利用度,并有助于避免过度的临床试验。专有药物之间活性药物成分相同的药物如美国食品和药物管理局(fda)所声明的,如果试验制剂和参比制剂之间的药物吸收速率和程度没有显着差异,则该制剂可以被认为是生物等效的。迄今为止,许多方法已被广泛用于ka的估计,一般大致可分为两种不同的类别: (i)隔室药代动力学(pk)模型方法,包括wagner-nelson方法(适用于单室pk模型)和 loo-riegelman方法(适用于两室pk模型);(ii)非区室pk模型方法,包括数值反卷积法和统计矩法。
4.除了吸收和消除阶段,药物遵循双室模型包含一个分布阶段,即药物从中央室分布到外围室,这不同于将身体视为均匀的单室模型组件(图1a、图2d)。在这种情况下,loo-riegelman 方法成为考虑分布阶段的经典方法,用于使用双室模型估计药物的ka,该方法需要pk参数,包括k
10
(一级消除速率常数)、k
12
(药物从中央隔室转移到外围隔室的一级速率常数)和k
21 (药物从外围隔室到中央隔室),均需要通过静脉注射相应药物来估计ka。数值反卷积方法计算药物的ka不受隔室模型的限制,但它需要静脉内和血管外给药的采样时间和间隔相同,因此,当使用loo-riegelman方法或数值反卷积方法时,也需要静脉内pk数据来估计ka。然而,由于人类志愿者的安全考虑,如果药物只能通过血管外途径给药,则很难确定药物的静脉内pk参数。统计矩法也可以应用于使用药物体内过程获得的整体随机变量的非隔室pk模型, ka是通过计算不同给药之间平均停留时间(mrt)的差异来估计的,以避免使用静脉内pk 数据,然而,许多因素影响使用统计矩法估计ka的准确性,例如检测低药物血浆浓度的精度,以及缺乏适当的数据来确定获得准确消除速率常数的终末相的对数线性(kt)。因此,静脉内 pk数据的缺陷或准确性差阻碍了使用双室模型估计药物的ka。
5.基于此,提供一种在无需静脉血药浓度数据的前提下,就能够满足对不同类型药物的药物吸收速率常数ka值进行准确估算的方法具有重要意义。
技术实现要素:
6.针对现有技术的不足,本发明提供了一种药物吸收速率常数的直接估测方法,该方法能够在不依赖于房室模型的吸收动力学数据的前提下,通过构建的直接估测模型直接估算获得 ka值。本发明不依赖于房室模型的直接估测模型估测的ka值准确度高,且无需静脉血药浓度数据,可满足对不同类型药物的药物吸收速率常数ka值估算,进而为药物制剂的体内吸收动力学解析及其ivivc的体内评价提供有价值的支持。
7.为实现上述目的,本发明所采用的技术方案具体如下所述:
8.一种药物吸收速率常数的直接估测方法,该方法包括如下步骤:
9.1)选取目标药物,并获取目标药物体内吸收特性的药动学参数。
10.2)定义目标药物吸收终点时间对应的药物处置率为最大表观处置速率常数。
11.3)根据最大表观处置速率常数建立不依赖于房室模型的直接估测模型,并采用该模型直接估算获得药物吸收速率常数ka。
12.作为优选,所述不依赖于房室模型的直接估测模型为:
[0013][0014]
式i中,t
max
为药物血浆浓度达峰时间,h。τ为药物吸收终点时间,h。k
max
为最大表观处置速率常数。
[0015]
作为优选,步骤1)具体为:将该目标药物进行血管外给药并采样,根据采样结果绘制实测血药浓度-时间曲线,获得药物体内吸收特性的药动学参数。其中血药浓度-时间曲线中血药浓度峰值对应的时间值即为t
max
。
[0016]
作为优选,对血药浓度-时间曲线进行求导获得导数曲线,根据导数曲线获得药物吸收终点时间τ,其中τ对应的导数曲线中的点即为最大表观处置速率常数k
max
。
[0017]
作为优选,所述不依赖于房室模型的直接估测模型构建过程如下:
[0018]
i)基于单室模型血管外给药的血药浓度计算公式(式ii),获得关于时间t的微分公式(式 iii):
[0019][0020][0021]
式ii-式iii中,f是生物利用度,x0是剂量,v是表观分布容积,k是消除速率常数。
[0022]
当药物血浆浓度达到最大药物血浆浓度c
max
时,式iii简化为式iv:
[0023]
[0024]
ii)对血药浓度-时间曲线进行求导获得导数曲线,根据导数曲线对k
max
、k、t
max
和τ进行拟合得到式v:
[0025][0026]
结合式iv和式v即得到式i:
[0027][0028]
作为优选,采用不依赖于房室模型的直接估测模型直接估算获得药物吸收速率常数ka为通过牛顿迭代法估算ka。
[0029]
作为优选,采用不依赖于房室模型的直接估测模型直接估算获得药物吸收速率常数ka为使用python软件包通过牛顿迭代法估算ka。
[0030]
作为优选,所述血管外给药为口服、肌肉注射或皮下注射、透皮给药、粘膜给药中的一种。
[0031]
作为优选,所述目标药物为满足双室模型的药物。
[0032]
作为优选,所述目标药物为醋酸阿比特龙片、阿昔洛韦混悬剂、阿奇霉素片、苯那普利胶囊、安非他酮片、坎地沙坦酯片、卡托普利片、塞来昔布胶囊、环丙沙星片、氯吡格雷片、达卡他韦片、多潘立酮片、屈他维林片、格列本脲片、氢氯噻嗪片、伊拉地平胶囊、伊曲康唑片、拉西地平片、盐酸乐卡地平片、左炔诺孕酮片、氯雷他定片、二甲双胍片、吗替麦考酚片、萘普生片、奥美沙坦酯片、磷酸奥司他韦胶囊、喹那普利片、瑞格列奈片、利匹韦林片、瑞舒伐他汀片、西洛多辛胶囊、辛伐他汀片、替米沙坦片、富马酸替诺福韦酯片、特比萘芬片、替格瑞洛片中的一种。
[0033]
在现有技术中,大多数药物在血管外给药后的体内表现与双室药代动力学(pk)模型拟合良好,但在没有静脉内pk数据的情况下,这些药物的吸收速率常数(ka)难以估算。对于现有技术的不足,针对性的开发了一种新方法,称为直接法,即通过提出定义最大表观处置速率常数(k
max
),在不使用静脉内pk数据的情况下估算药物的ka值。同时还通过使用设置参数和临床数据的方式来确定本发明直接法估算的ka的准确性。结果表明,直接法估计ka的绝对相对误差(re)明显低于loo-riegelman法和统计矩法对集合数据的绝对相对误差(re)。进一步地还通过对替米沙坦、坎地沙坦酯和富马酸替诺福韦酯的人体pk研究表明,基于ka值与反映药物吸收特性的其他体内pk参数(t
max
、c
max
和c
max
/auc
0-t
)之间的良好相关性,这些药物的ka值均能够通过直接法进行准确的估算。这种新方法有望为pk评估和ivivc建立提供有价值的支持。
[0034]
在本发明中,通常血管外给药的单室模型的血浆浓度(c)和ka具有以下关系(式ii):
[0035][0036]
其中,f是生物利用度,x0是剂量,v是表观分布容积,k是消除速率常数。则关于时间t的微分公式为(式iii):
[0037][0038]
当药物血浆浓度达到峰值c
max
(例如)时,式iii简化为式iv:
[0039][0040]
在本发明中,在不考虑pk模型的情况下,血药浓度-时间曲线可以看作由两部分组成:一级速率增加曲线和一级速率减小曲线。满足基本公式c=ae-kt-be-kat
,其中k表示单室模型中的消除速率常数或双室模型中由于分布(k
12
)和消除(k
10
)而从中央隔室中取出的药物的总去除速率常数。因此,在式iv中的k被替代方法的“k
12
k
10”替代之后,估计了与双室模型拟合的药物的ka。与loo-riegelman方法和统计矩方法相比,该替代方法具有出色的准确性和便利性。然而,替代方法也需要静脉内pk数据来计算k
10
和k
12
。因此,在双室模型中研究一种新的pk参数以替换k(在式iv),是在没有静脉内pk数据的情况下,估计ka的有效方法之一。
[0041]
在本发明中,通过定义了一个新参数,称为最大表观处置速率常数(k
max
),并以该定义的k
max
开发了一种用于估计ka的新方法(称为直接法)。在从前发表的报告中,确定这些参数的关系和范围后,通过将k
12
、k
21
、k
10
值分别设置为高、中和低水平来研究直接法估计的 ka的准确性。还将直接法估计的ka的精确度与使用loo-riegelman法和统计矩法确定的相应的ka的精确度进行比较。本发明选择了三种不同配方的模型药物替米沙坦(tms)、坎地沙坦酯(csc)和富马酸替诺福韦酯(tdf),并在人体中评估了它们的pk参数。然后采用直接法估计三种模型药物的ka,从中建立它们估计的ka值与反映药物体内吸收特性的其他pk 参数(例如:t
max
、c
max
和c
max
/auc
0-t
)之间的相关性,通过分析这些相关性以验证直接方法所估计的药物ka的准确性并衡量其应用范围。
[0042]
在本发明中,与k值不变的单室模型不同(图1b),用双室模型固定良好的药物血药浓度-时间曲线分为三个阶段:吸收相、吸收后相和处置相(处置相=分布相 消除相)(图2e)。其中t
max
之前的曲线部分代表吸收阶段,在此期间药物血浆浓度的增加速率高于其处置速率。而t
max
之后的曲线部分是吸收后阶段,在此期间药物的处置速率高于其吸收速率。此后,药物处置率逐渐下降,直至达到一个不变的终端淘汰过程。在后吸收阶段的结束时间(τ),药物的吸收阶段已经完成,因此只剩下药物的处置阶段。使得它在τ后的第一个时间间隔内呈现出最高的药物处置率(即k
max
)(图2e)。此外,血药浓度-时间曲线的对数导数反映了药物浓度的实时下降速率(即t
max
后对数pk曲线的斜率),该速率逐渐增加,最后保持恒定速率 (k)。对于单室模型,由于在t
max
之后存在后吸收阶段(图1c),而药物浓度下降的速率具有增加、减少和恒定的连续过程,这使得在双室模型的τ处呈现k
max
(图2f)。
[0043]
在本发明中,通过将单室模型和双室模型中的ka、x0、f和v设置为相同的值,以及 k=k
12
k
10
。发现两条模拟的血药浓度-时间曲线的吸收阶段几乎重叠(图3g)。通过去卷积后的吸收曲线也重叠(图3h),在τ时间点药物吸收基本完成,并对应于k
max
。对血药浓度-时间曲线求导后发现k
ma
x和k不相等,并且k
max
的值总是小于k的值。进一步地,还发现当按比例进行缩放后,k
max
、k、t
max
和τ的值具有以下关系(图3i~j):
[0044][0045]
转化后获得式v:
[0046][0047]
结合式iv和式v即得到不依赖于房室模型的直接估测模型式i:
[0048][0049]
在这种情况下,t
max
值是直接从血药浓度-时间曲线上获得的,而k
max
和τ是从血管外给药后双室模型的血药浓度-时间曲线的对数获得的。通过使用python软件包(版本3.6.7)进行牛顿迭代法估算ka。因此,直接法不需要测量药物的静脉内浓度的优点。
[0050]
与现有技术相比较,本发明的有益技术效果如下:
[0051]
1:本发明提供了一种直接估算ka的方法,通过提出定义k
max
为最大表观处置速率常数,在不依赖房室模型的吸收动力学模型解决传统的方法估算ka值的弊端,创造性地提出了不依赖房室模型、仅与药时曲线特征有关的体内吸收动力学模型(式i),大大提高了解析估算ka 值的准确度。
[0052]
2:本发明通过直接估测模型解析的ka值准确性高,适用范围广泛,且能为药物制剂的体内吸收动力学解析及其体内外相关性(ivivc)的体内评价提供有价值的支持。
附图说明
[0053]
图1为单室模型及其血药浓度-时间曲线图组。
[0054]
图2为双室模型及其血药浓度-时间曲线图组。
[0055]
图3为单室模型和双室模型设定数值的模拟药物浓度-时间曲线及去卷积后吸收曲线图组。
[0056]
图4为39组设定参数的血药浓度-时间曲线图组。
[0057]
图5(a)为直接法、统计矩方法以及loo-riegelman方法计算的re的绝对值对比图。
[0058]
图5(b)为直接法、统计矩方法以及loo-riegelman方法计算的re的中值对比图。
[0059]
图6(a)为直接法、统计矩方法以及loo-riegelman方法估算的ka受k
12
影响的变化图。
[0060]
图6(b)为直接法、统计矩方法以及loo-riegelman方法估算的ka受k
21
影响的变化图。
[0061]
图6(c)为直接法、统计矩方法以及loo-riegelman方法估算的ka受k
10
影响的变化图。
[0062]
图7(a)为tms片剂的平均血药浓度-时间曲线图。
[0063]
图7(b)为csc片剂的平均血药浓度-时间曲线图。
[0064]
图7(c)为tdf片剂的平均血药浓度-时间曲线图。
[0065]
图8为tms片剂的药物制剂分别采用直接法、统计矩方法以及loo-riegelman方法
的平均吸收分数时间曲线图组。
[0066]
图9为csc片剂的药物制剂分别采用直接法和统计矩方法的平均吸收分数时间曲线图组。
[0067]
图10为tdf片剂的药物制剂分别采用直接法和统计矩方法的平均吸收分数时间曲线图组。
[0068]
附图释义:two-compartment model:双室模型;one-compartment model:单室模型;plasmaconcentration:血药浓度;time:时间曲线;absorption phase:吸收相;post-absorption phase:吸收后相;elimination:消除相;disposition phase:处置相;distribution:分布;elimination:消除;central compartment:中央室;peripheral compartment:周边室;deconvolution:逆卷积分;derivative of the logarthm of pk profile after t
max
:t
max
后对数pk曲线的导数;value:值; concentration:浓度;absolute re%:re的绝对值%;median re%:re的中值%;methods:方法;plasma concentration of tms:tms血药浓度;plasma concentration of candesartan:坎地沙坦的血药浓度;plasma concentration of tenofovir:替诺福韦的血药浓度。
具体实施方式
[0069]
下面对本发明的技术方案进行举例说明,本发明请求保护的范围包括但不限于以下实施例。
[0070]
实施例1
[0071]
通过设置参数验证直接法
[0072]
1.1参数设置与剂型判断:
[0073]
采用getdata graph digitizer软件(2.25版本,http://www.getdata-graph-digitizer.com)从现有技术的文献中获取36个满足双室模型药物的血药浓度数据。然后使用winnonlin软件 (8.2版本,certara公司,usa)初步计算这些药物的ka、k
12
、k
21
和k
10
值,并比较各参数间的关系。将所获得的36个药物的ka、k
12
、k
21
和k
10
值按降序排序,再将这些参数的前1/3、中1/3和后1/3(n=12)的平均值分别设置为高、中、低数值水平。每个参数的不同级别间随机组合,再将这些参数(ka、k
12
、k
21
和k
10
)带入公式1~公式3,进而获得不同时间点(间隔0.1h)的血药浓度,并绘制药时曲线(血药浓度-时间曲线):
[0074][0075][0076][0077]
其中x0、f和vc被随机设置为固定值(例如:x0=2200μg,f=1,vc=10l)。公式1中的α和β为变量,分别表示分布相混合一级速率常数和消除相混合一级速率常数,分别由公式2 和公式3确定。
[0078]
此外,使用公式4和公式5计算aic值,评估血药浓度-时间曲线的隔室模型:
[0079]
aic=n
·
lnre 2p......(4)
[0080][0081]
其中n是实验组数,re是加权残差平方和,p是模型参数个数,wi代表权重系数,ci是实验药物血浆浓度,是估算的药物血浆浓度。
[0082]
软件估算的36种ir剂型药物在人体口服给药后两室pk模型中的ka、k
10
、k
12
和k
21
值 (p《0.001vsk
12
,k
21
和k
10
;p《0.01vsk
21
;p《0.05vsk
21
)。
[0083]
表1.aic计算结果
[0084]
[0085][0086]
aic1:单室模型的aic值;aic2:双室模型的aic值;na:不适用。
[0087]
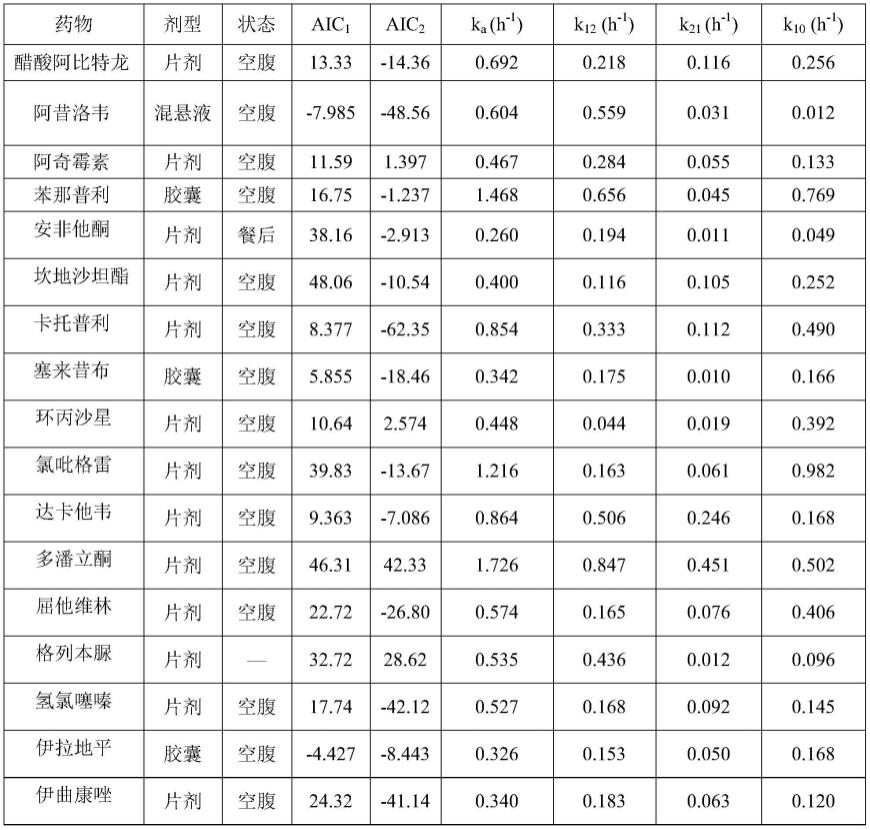
从文献中获取了36个不同速释(ir)剂型的人体内血药浓度数据,采用winnonlin软件计算各药物的aic值。结果如表1所示,所有药物的aic2值(双室模型)均小于aic1值(单室模型),表明36个药物的体内过程均符合双室模型。再通过winnonlin软件初步估算了各药物的ka(0.210~1.826h-1
)、k
12
(0.044~0.847h-1
)、k
21
(0.010~0.451h-1
)和k
10
(0.012~1.003h-1
) 范围。通过对比各参数间的关系发现,所有药物的k
12
和k
10
值之和均小于ka值(即ka》k
12
k
10
),各药物的ka值均大于k
12
值,且k
12
值均高于k
21
值(即ka》k
12
》k
21
)。除少数药物(如阿昔洛韦、达卡他韦和左炔诺孕酮)外,k
10
值均显著高于k
21
(p《0.05),该结果为满足双室模型药物的ka、k
10
、k
12
和k
21
参数设定提供重要依据。
[0088]
1.2采用直接法估算ka:
[0089]
t
max
是从设定参数的血药浓度-时间曲线的数据中获得的。k
max
是从τ时间点之后的
第一个时间间隔的血药浓度-时间曲线的对数斜率拟合的。然后通过直接法(公式(i))估算ka的值。ka的准确度是通过比较公式(i)中估算的ka来计算的。以下使用公式6得到ka的设置值(即,真正的ka):
[0090][0091][0092]
1.3采用loo-riegelman方法估算ka:
[0093]
设置k
12
、k
21
和k
10
值用于通过loo-riegelman方法估算ka。简而言之,ka由公式7进行计算:
[0094]
ln(1-f
abs
)=-kat b......(7)
[0095]
其中体内吸收分数(fabs)由公式8获得。:
[0096][0097]
公式8中的值由公式9计算获得:
[0098][0099]
其中,表示药物量分别在时间(xa)
t
和(xa)
∞
时进入体循环。(x
p
)
t
表示在时间t进入外围隔室的药物量。此外,δc和δt分别代表两个连续样本之间的血浆浓度和时间差异。
[0100]
1.4采用统计矩法估算ka:
[0101]
将设定参数的血药浓度-时间数据用统计矩法拟合得到的ka值与直接法拟合得到的ka值进行比较。为进行这种比较而执行的计算如公式10所示:
[0102][0103]
在公式10中,mat是平均吸收时间,mrt是血管外给药后的平均停留时间。k
t
是终末期的消除速率常数。使用梯形法计算auc(血药浓度-时间曲线下面积)。aumc代表力矩曲线下的面积,其由下述公式11计算:
[0104][0105]
其中,ci、c
i 1
和cn分别表示ti、t
i 1
和tn时间点的药物血浆浓度。
[0106]
为考察直接法的准确度和灵敏度,ka、k
12
、k
21
、k
10
的高、中、低值根据以往的报道(表 1)设定。ka的设定值为1.098、0.603、0.375h-1
;k
12
的设定值为0.525、0.211、0.133h-1
;k
21
的设定值为0.176、0.067、0.025h-1
;k10的设定值分别为0.571、0.271和0.100h-1
(表2)。
根据ka、k
12
、k
21
、k
10
之间的关系(ka》k
12
k
10
,ka》k
12
》k
21
),最后通过组合ka、k
12
、k
21
和 k
10
的值来呈现39组(如表2所示),所有组均满足双室模型(aic1》aic2)。t
max
、k
max
和τ的值是从相应组的血药浓度-时间曲线获得的(图4),其中t
max
随着ka的降低而增加。然后通过直接法、loo-riegelman法和统计矩法估算ka的值。直接法估算的ka的re与集合k
a (即真实ka)相比既有正值又有负值,大多数组的值小于20%。然而,使用loo-riegelman 方法,所有re值都是正的,其中估算的ka》真实的ka。相反,使用统计矩法,大部分re值为负,因此估算的ka《真实的ka。
[0107]
表2.采用设定值通过不同方法估算的ka值的准确性(39组)
[0108]
[0109][0110]
aic1:单室模型的aic值;aic2:双室模型的aic值;dm:直接法;l-r:loo-riegelman方法;stm:统计矩法;na:不适用;mat:阴性。
[0111]
计算re的绝对值并显示在图5(a)中。来自直接法的ka的绝对re显着小于来自统计矩方法(p《0.01)或loo-riegelman方法(p《0.001)的re。loo-riegelman方法的ka绝对re明显小于统计矩方法的re(p《0.01)。图5(b)表明,直接法估算的ka中值re(
–
4.98%)优于loo-riegelman法(21.5%)和统计矩法(
–
65.9%)估算的re。此外,与loo-riegelman法和统计矩法相比,使用直接法估算ka的准确性不受k
12
、k
21
和k
10
变化的影响,这也表现出优异的准确性(图6(a)-6(c))。因此,本发明的直接法更准确,不需要通过静脉内pk测量来确定k
12
、k
21
和k
10
。
[0112]
实施例2
[0113]
通过临床数据验证直接法
[0114]
2.1模型药物临床资料:
[0115]
三种模型药物替米沙坦(tms)、坎地沙坦酯(csc)和富马酸替诺福韦酯(tdf)的药物血浆浓度是从使用健康人类志愿者的pk研究中获得的。临床研究按照赫尔辛基宣言进行,方案经中国食品药品监督管理总局(cfda)和中南大学湘雅药学院机构研究伦理委员会批准(项目代码:2020006)。所有登记的志愿者都被充分告知了临床研究的方案,并同意他们参与。pk研究采用随机、开放标签、单剂量设计进行,以比较分别口服含有tms、csc 和tdf的不同制剂后的pk参数。其中,不同速释(ir)剂型的片剂剂型包括tms(f
m1
、 f
m2
,规格:80mg)、csc(f
c1
、f
c2
,规格:4mg)和tdf(f
d1
、f
d2
,规格:30mg),分别由三个不同的制药公司友情提供。
[0116]
tms片剂的pk研究是在26名健康志愿者中使用禁食状态和双向交叉设计进行的,其中包括治疗之间的7天清除期。在给药前(0小时)和分别在给予f
m1
或f
m2
片剂后的0.17h, 0.33h,0.5h,0.75h,1h,1.25h,1.5h,2h,2.5h,3h,4h,6h,8h,10h,12h,24h,48h, 72h和96h采样,并将血样收集在含有肝素的真空采血管中。
[0117]
csc片剂的pk研究在24名志愿者中进行,使用空腹状态、双向交叉设计,治疗之间有 7天的清除期。在给药前(0小时)和分别在给予f
c1
或f
c2
片剂后0.33h,0.67h,1h,1.33h, 1.67h,2h,2.33h,2.67h,3h,4h,6h,8h,12h,24h和48h采样,并将血样收集在含有肝素的真空采血管中。
[0118]
tdf片剂的pk研究在24名志愿者中进行,采用空腹状态和餐后状态的双向交叉(餐后状态包括高脂肪膳食,营养成分为522kcal脂肪、288kcal碳水化合物、149kcal蛋白质和959 卡路里的总热量)。使用tdf片剂的研究以治疗之间的7天清除期为特征。在给药前(0小时) 和分别在f
d1
或f
d2
片剂给药后0.25h,0.5h,1h,1.5h,2h,2.5h,3h,3.5h,4h,5h,6h, 8h,10h,12h,24h,36h和48h采样,并将血样收集在含有肝素的真空采血管中。
[0119]
对于所有血液样本,在以3500rpm离心10分钟后分离血浆。然后将血浆样品储存在
–
70℃冰箱中保存,直到通过高效液相色谱串联质谱(hplc-ms/ms,agilent,usa)进行分析。
[0120]
从胃肠道吸收后,csc和tdf在血浆中分别迅速水解为坎地沙坦和替诺福韦。使用winnonlin软件包计算tms、坎地沙坦和替诺福韦的pk参数,包括c
max
、t
max
、auc
0-t
、 auc
0-∞
和消除半衰期(t
1/2
)。所有数据均表示为平均值
±
标准差(sd)。所有统计分析均由spss 软件包(25.0版,spss inc.,usa)进行,p《0.05被认为具有统计学意义的差异。
[0121]
2.1直接法的验证
[0122]
tms、csc和tdf的k
max
和τ值通过计算血药浓度-时间曲线的对数获得。tms、csc 和tdf的ka通过直接法(公式i)、统计矩法(公式10)和loo-riegelman法(公式7)估算。进行pearson相关分析(spss,版本25.0,spss inc.,usa)以评估ka值与其他反映药物体内吸收特性的pk参数(t
max
、c
max
和c
max
/auc
0-t
)之间的关系。此外,使用公式12拟合吸收率与时间的曲线:
[0123]fabs
=[1-exp(-kat)]*100%......(12)
[0124]
tms、坎地沙坦和替诺福韦的平均血药浓度-时间曲线是从人体pk评估中获得的
(图7 (a)~7(c))。其pk参数列于表3。
[0125]
单剂量的tms(n=26),csc(n=24)和tdf片剂(n=24)分别在空腹或/和餐后状态给药后 tms、坎地沙坦(csc的代谢物)、替诺福韦(tdf的代谢物)的pk参数。所有数据均为平均值
±
sd,p《0.05对比空腹状态下的相同剂型。
[0126]
表3.人体pk研究参数
[0127][0128]
在口服给药后0.5-3小时内,f
m2
的平均血浆浓度高于f
m1
(图7(a)),f
m2
的c
max
高于f
m1
(表3)。总体而言,f
c1
和f
c2
的血药浓度-时间曲线(图7(b))和pk参数(表3) 相似。对于f
d1
和f
d2
(p《0.05,图7(c),表3),餐后状态下替诺福韦的c
max
明显低于空腹状态下替诺福韦的c
max
,而餐后状态下替诺福韦的t
max
状态也大于空腹状态下替诺福韦的t
max
(p《0.05for f
d2
)。具有不同t
max
值(0.5-4h)的三种模型药物代表了ir剂型的低、中、高吸收率。
[0129]
tms、csc、tdf片剂的ka值通过不同的方法估算。tms的静脉内pk参数来自先前发表的报告,并用于通过loo-riegelman方法估算ka。然而,很难获得静脉给药后csc、tdf 及其各自代谢物(坎地沙坦、替诺福韦)的体内数据。如表4所示,直接法得到的f
m2
的ka值高于同法得到的f
m1
的ka。这与使用loo-riegelman方法对ka的估算是一致的,但与使用统计矩方法对ka的估算相反。f
c1
的直接法估算ka与f
c2
的估算ka相似,而f
c1
的统计矩法估算的ka高于f
c2
的估算ka。空腹状态下f
d1
和f
d2
的估算ka均高于餐后状态下f
d1
和f
d2
的估算ka(对于f
d2
,p《0.05)。空腹状态下f
d1
和f
d2
的k
max
也高于餐后状态下f
d1
和f
d2
的 k
max
(p《0.05)。此外,f
d1
的ka值与f
d2
的ka值在相同状态下使用直接法是一致的。这与统计矩法相反,后者产生的f
d1
的ka高于f
d2
。
[0130]
通过不同方法对餐后或/和空腹状态下的tms、csc、tdf片剂的ka进行估算。所有数据均为平均值
±
sd,p《0.05,使用直接法在空腹状态下的相同剂型。
[0131]
表4:通过不同方法对ka进行估算
[0132][0133]
na:不适用,因为它没有静脉内pk数据。
[0134]
tms片剂的平均吸收分数时间曲线表明,使用直接法和loo-riegelman法(图8a1、8b1) 在前4小时内f
m2
的吸收分数快于f
m1
,这与tms的血浆浓度-时间曲线平均值(图7(a)) 和c
max
值(表3)一致。然而,使用统计矩法(图8c1),f
m1
和f
m2
的吸收曲线几乎重叠,这与它们的体内实验数据不一致。基于pearson相关分析,直接法估算的ka值与c
max
和 c
max
/auc
0-t
均呈正相关(相关系数(r)》0.4,p《0.01,图8a2-8a3),与t
max
负相关(r=-0.858, p《0.001,图8a4)。然而,使用loo-riegelman方法(图8b2-8b4)和统计矩法(图8c2-8c4) 估算的ka与这些参数几乎没有相关性(p》0.1)。
[0135]fc1
和f
c2
之间估算的ka的相似性导致使用直接法几乎重叠的吸收分数时间曲线(图 9d1)。估算的ka值与c
max
和c
max
/auc
0-t
呈正相关(p《0.01,图9d2-9d3),与t
max
负相关 (p《0.001,图9d4)。然而。使用统计矩方法(图9e1),这两个曲线并不相似,这与它们的体内性能不一致(图7(b))。通过统计矩方法估计的ka也与c
max
和c
max
/auc
0-t
几乎没有相关性(图9e2-93),而与t
max
呈正相关(图9e4)。
[0136]
通过直接法(图10f1)和统计矩法(图10g1)估算ka值后,获得了tdf片剂的平均吸收分数时间曲线。使用直接法对tdf的吸收估算,两种配方(f
d1
和f
d2
)中tdf的吸收在空腹状态下均高于餐后状态下。在餐后和空腹状态下,f
d1
和f
d2
的估算ka值与c
max
、 c
max
/auc
0-t
和t
max
的相应平均值密切相关(r》0.96,p《0.05,图10f2-10f4)。然而,吸收曲线与使用统计矩法(图10g1)的体内浓度数据(图7(c))不一致。此外,通过统计矩法估算的ka与c
max
、c
max
/auc
0-t
、t
max
几乎没有相关性(p》0.6,图10g2-10g4)。
[0137]
结果讨论
[0138]
双室模型药物的ka、k
12
、k
21
和k
10
值因其理化性质和剂型而异,但这些参数之间的关系至今未见报道。药物速释制剂的估算ka、k
12
、k
21
和k
10
值的准确性优于缓释制剂的相应
准确性,因为前者受体内溶出速率的影响较小。在这些情况下,采用具有不同t
max
(0.75
–
4.0h)和 t1
/2
(1.2
–
52.8h)值且满足双室模型的36种速释(ir)剂型来估算ka、k
12
、k
21
和k
10
值(表1) 用于调查这些参数之间的关系。理论上,k
12
的值应高于k
21
(k
12
》k
21
),因为药物从中央隔室到外围隔室的分布是动态的。同时,药物的吸收率需要大于分布和消除率之和(ka》k
12
k
10
),以保证血管外给药后血浆中的药物浓度可以测定。阐明这些参数之间的关系可以避免在设置数据以研究直接法时出现的任何空白。然而,使用winnonlin软件(内置残差法)仅对36种速释(ir)剂型的ka、k
12
、k
21
和k
10
值进行了初步量化以观察它们的关系。不出所料,winnonlin 软件估计的tms、csc和tdf的ka与直接法计算的ka值存在差异(表4)。
[0139]
所有设置组的t
max
范围为0.5-4.0h(表2),这代表了大多数ir剂型在实践中的体内性能。直接法估计的ka明显受k
max
、t
max
和τ(式i)的影响,而k
max
和t
max
(或τ)之间的负相关可以保证估算的ka准确且独立于变化在k
12
、k
21
和k
10
(图6(a)-6(c))中。作为一种非隔室方法,统计矩法应该对隔室参数(即k
12
、k
21
和k
10
)的变化不敏感。然而,通过统计矩法估算的ka的大部分值处于较低水平(表2),因为k
t
的值较小是从终端采样点获得的。当使用loo-riegelman方法时,根据公式8和公式9,ka的估算值无疑且敏感地受到k
12
、k
21
和k
10
(图6(a)-6(c))影响。然而,ka的所有估算值都高于ka的真实值,这可能归因于未吸收部分(1-f
abs
%)中时间点数量的差异通过线性回归拟合获得。此外,使用loo-riegelman 法估算的ka的平均绝对re值较大,因为存在一些异常值(re》100%),对拟合精度产生负面影响,但它也具有比统计矩更好的估算精度方法(图5(a)-5(b))。
[0140]
选择t
max
(0.5-4h)不同的三种模型药物,在实践中探索直接法的准确性和范围。这些药物的经验ka值无法从报告的研究中获得。因此,研究了吸收率和pk数据之间的关系,以间接验证直接法的准确性。一般来说,药物的高吸收率导致c
max
大而t
max
短。c
max
和c
max
/auc
0-t
的值代表药物的体内暴露量,这也与ka的值有关.通过直接法估计的三种模型药物的ka值与 tms(图8a2-8a3)、csc(图9d2-9d3)和tdf(图10f2-10f3)的体内暴露呈正相关,其中可能有利于预测不同制剂的体内暴露。在ka和t
max
之间观察到负相关(图8a4、9d4、 10f4),这与以前的文献结果一致。然而,loo-riegelman方法(仅用于tms)和统计矩方法都未能建立估算的ka与其c
max
、c
max
/auc
0-t
和t
max
值之间的相关性。统计矩法估算的csc 的ka与t
max
呈正相关,这与文献先例相反。
[0141]
药物的pk参数,包括c
max
、auc
0-t
、t
max
和ka,通常受高脂肪食物的影响。在本研究中,相比空腹状态下,餐后状态下tdf具有降低的ka和c
max
值以及延长的t
max
值。之前的文献报告在餐后和空腹状态之间观察到tdf的相似体内结果。使用直接法(表4)在餐后和空腹状态之间观察到tdf的估算ka差异,并且它也与c
max
、c
max
/auc
0-t
和t
max
呈线性相关。相反,统计矩法未能在餐后和空腹状态之间产生估算的ka差异,并且在估算的ka和它们的 c
max
、c
max
/auc
0-t
和t
max
值之间没有观察到相关性。因此,这些结果证实了直接法对于估算与pk评估相关的应用的ka是灵敏且准确的。
[0142]
尽管先前已经证明口服给药后药物的吸收在t
max
后的有限时间内终止,但确切的终点尚不清楚。在本研究中,τ代表pk曲线中吸收后阶段的终点,在该终点,吸收过程完成。tms、csc、tdf片剂的τ值是在餐后和/或空腹状态下获得的(表4)。这些药物的f
abs
平均值在τ的平均值处超过90%(图8a1、9d1、10f1),验证了直接法的推论。
[0143]
由于k
max
、τ和t
max
的准确性也极大地影响了ka的估计,因此可能需要pk研究中足够
的采样点来获得准确的k
max
、τ和t
max
。本研究将三种模型药物在人体pk研究的采样点设计为常规采样点(如0.17h、0.33h、0.5h、1h
……
),而不是间隔为设置数据为0.1小时。结果表明,常规点对ka的计算没有显著影响,表明直接法在实际应用中估算药物吸收率是高度可行的。
[0144]
综上所述,在本发明中,使用公式(i)
[0145]
的双室模型来估算药物的ka,其中t
max
、k
max
和τ的值很容易从血管外给药后的血浆浓度-时间曲线中获得。与loo-riegelman方法和统计矩法相比,使用设定数据的直接法估算的ka具有令人满意的准确性。另一方面,三种模型药物(tms、csc、tdf)的估算ka值采用直接法估算,与相应的pk曲线一致。根据这些计算,在ka值和反映药物体内吸收的其他pk参数之间建立了良好的相关性。这些结果证实了估算药物吸收率的直接法的准确性,在无法获得静脉内pk数据的应用中是有益的。这种直接法有望为 pk评估和ivivc建立提供有价值的支持。
[0146]
在本发明中,关于体内吸收速率常数的解析方法(python迭代法程序)如下:
[0147]
[0148]
[0149]
[0150]
[0151]
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。