1.本发明涉及用于制备异烟肼的多步连续流合成方法,具体而言,但不限于产率超过90%的制备异烟肼的连续流合成方法。
2.发明背景
3.现有技术中描述了许多制备异烟肼的方法和合成路线。
4.然而,用于生产这些化合物的现有合成方法都是基于标准搅拌间歇反应器类型的工艺,其中使用了大量有机溶剂。此外,这些工艺通常在工艺的每一步分离工艺中间体,从而进一步增加溶剂的使用。因此,以这种方式生产的原料药成本相对较高,主要是因为对有机溶剂的依赖,以及由于分离中间体而导致的固有工艺效率低下。
5.微反应器技术(mrt),最近被称为“流动化学”,是一种新兴技术,使从事研究和开发的人员能够利用连续流快速筛选反应,从而确定适合在生产水平上使用的反应条件。此外,除了使用常规反应方法外,与使用小反应器体积相关的固有安全性允许用户使用之前认为在生产环境中使用过于危险的反应条件;例如极端反应条件或使用/产生“危险”化合物。因此,通过使用这项技术,化学家可获得的反应类型增加了。迄今为止,还没有基于流动化学方法生产异烟肼的工艺。
6.因此,需要一种改进的制备异烟肼的方法。具体而言,需要一种用于制备异烟肼的全连续多步流动合成方法,该方法优选产生超过90%的异烟肼产率。
7.发明概述
8.根据本发明的第一方面,提供了一种用于制备式1的异烟酰肼的多步连续流合成方法,其适于产生大于约90%的产率,
[0009][0010]
包括以下步骤:
[0011]
a)将式12的4-氰基吡啶
[0012][0013]
与naoh在约90℃至约105℃的温度下以约1:0.1至约1:0.7的4-氰基吡啶:naoh的摩尔比反应以产生式13的中间体异烟酰胺
[0014][0015]
b)使式13的中间体异烟酰胺与水合肼在约100℃至约120℃的温度下以约1:1.75至约1:2.50的4-氰基吡啶:水合肼的摩尔比反应,
[0016]
其中,步骤a)中的反应的停留时间为约7至约32分钟,且步骤b)中的反应的停留时间为约10至约25分钟。
[0017]
在一个实施方案中,在步骤a)中,将4-氰基吡啶溶解在水和醇的混合物中,包括甲醇。
[0018]
在优选的实施方案中,水和醇混合物的醇水比约为7:3。
[0019]
在一个实施方案中,在步骤a)中,4-氰基吡啶与naoh的摩尔比为约1:0.15至约1:0.6,或约1:0.15至约1:0.4。
[0020]
在优选的实施方案中,在步骤a)中,4-氰基吡啶与naoh的摩尔比约为1:0.2。
[0021]
在一个实施方案中,在步骤a)中,温度为约90℃至约100℃,或约95℃至约100℃。
[0022]
在优选的实施方案中,在步骤a)中,温度约为95℃。
[0023]
在优选的实施方案中,在步骤a)中,停留时间约为10到15分钟。
[0024]
在特别优选的实施方案中,在步骤a)中,停留时间约为10分钟。
[0025]
在一个实施方案中,在步骤b)中,4-氰基吡啶与水合肼的摩尔比为约1:1.75至约1:2.25。
[0026]
在优选的实施方案中,在步骤b)中,4-氰基吡啶与水合肼的摩尔比约为1:2.0。
[0027]
在另一个实施方案中,在步骤b)中,所述温度为约100℃至约115℃,或约105℃至约115℃。
[0028]
在优选的实施方案中,在步骤b)中,所述温度约为105℃。
[0029]
在优选的实施方案中,在步骤b)中,停留时间为约10至约20分钟。
[0030]
在特别优选的实施方案中,在步骤b)中,停留时间约为10分钟。
[0031]
在优选的实施方案中,由4-氰基吡啶制备异烟酰肼的多步连续流合成法总产率大于约92%、大于约94%或大于约95%。
[0032]
在特别优选的实施方案中,由4-氰基吡啶制备异烟酰肼的多步连续流合成法总产率为约96%。
[0033]
附图简述
[0034]
现在将参考以下非限制性实施方案和附图更详细地描述本发明,其中:
[0035]
图1示出了本发明的一般合成方法的示意图;
[0036]
图2示出了用于研究作为多步连续工艺制备异烟酰肼1的装置的示意图;
[0037]
图3示出了通过4-氰基吡啶水解合成异烟酰胺的连续流装置;
[0038]
图4示出了氢氧化钠相对摩尔百分比对转化为13和形成副产物15的影响;
[0039]
图5示出了温度对转化13和形成副产物15的影响;
[0040]
图6示出了停留时间对12转化为13的影响;和
[0041]
图7示出了总停留时间对转化为1的影响,其中转化为13的反应的停留时间设定为
10分钟。
[0042]
优选实施方案详述
[0043]
下面将参考附图更全面地描述本发明,其中示出了本发明的一些非限制性实施方案。
[0044]
下文所述的本发明不应被解释为仅限于所公开的特定实施方案,稍作修改和其他实施方案包括在本发明范围内。
[0045]
尽管本文使用了特定术语,但它们仅用于一般性和描述性的意义,而不是为了限制的目的。
[0046]
如本文所用,在本说明书和所附权利要求书中,单数形式“一个/种(a)”、“一个/种(an)”和“该/所述(the)”包括复数形式,除非上下文另有明确指示。
[0047]
本文使用的术语和措辞是出于描述的目的,不应被视为限制性的。本文使用的术语“包含”、“含有”、“具有”、“包括”及其变体旨在涵盖其后列出的项目、其等同物以及附加项目。
[0048]
在本说明书中使用时,术语“全连续流合成法”、“多步连续流合成法”和“全连续多步流动合成法”可互换使用并且应理解为表示流动合成方法,即使用微反应器技术的方法,包括多个步骤,其中该方法的最终产物是在不分离任何中间体的情况下获得的。
[0049]
本发明提供了一种以全连续流合成法制备异烟肼的方法。图1示出了本发明用于制备异烟肼的一个实施方案的一般合成方法和合成步骤的示意图。
[0050]
图1所示的异烟肼连续流合成方法是使用微反应器技术进行的两步反应。通常,转化为异烟肼的第一个反应是在第一微反应器或合适的微反应器装置的第一部分中水解4-氰基吡啶12以产生异烟酰胺13。异烟酰胺13在第二微反应器或合适的微反应器装置的第二部分中与水合肼反应,该装置与第一微反应器或微反应器的第一部分持续流体连通。
[0051]
用于实施本发明方法的微反应器装置的一个示例如图2所示。从图2可以看出,可以通过使用两个注射泵、三个注射器和各种反应器板来构建用于实施本发明的连续流反应装置。该系统可包括单向流止回阀和背压调节器。反应器板的温度可以例如通过使用油浴或以本领域已知的任何其他方式进行控制。本领域普通技术人员将理解,可以修改如上所述的实验设置的各种细节,从而得出进一步的实施方案,但是这些实施方案将保持在本发明的范围内。
[0052]
试剂和溶剂的相容性在流动系统中尤其重要,以确保产物和中间体保持在溶液中。不正确的溶剂、试剂比例、试剂浓度、反应温度和其他反应条件会导致试剂沉淀。沉淀不仅会影响反应效率,还会导致系统堵塞,进而导致系统压力升高。系统堵塞将导致代价高昂的系统停止运行,以及反应的整体低效。
[0053]
现在将参考以下非限制性实验实施例和分析数据更详细地描述根据本发明的方法的各个合成步骤。
[0054]
实验参数
[0055]
所有使用的化学品均购自merck,sigma aldrich和industrial analytica。它们按原样使用,无需进一步纯化。
[0056]
采用薄层色谱法(tlc)监测反应进程。以己烷和乙酸乙酯(80:20v/v)为流动相,在层厚250μm e.merck硅胶板(60f-254)上进行tlc。斑点的检测通过使用camag紫外检测器柜
在254nm处完成。化合物的纯度通过tlc板上的单个斑点确定。
[0057]
使用bruker光谱仪(bruker ultrashield tm 400plus)记录1h和
13
c核磁共振(nmr)光谱,该光谱仪在400mhz下用于氢谱和100mhz下用于碳谱。使用dmso-d6(2.62ppm)中残留1h化学位移(用作内标)校准光谱。所有光谱的化学位移值均以百万分之分数(ppm)给出。
[0058]
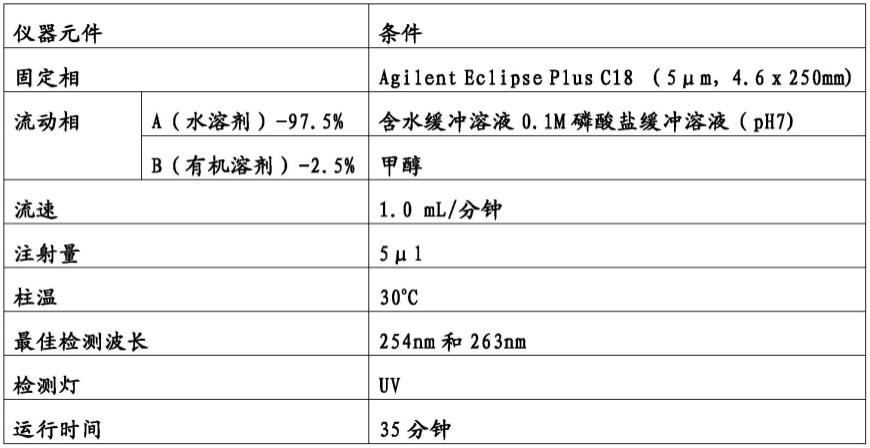
通过分析型高效液相色谱法(hplc)测定4-氰基吡啶12、异烟酰胺13、异烟酰肼1和异烟酸15。使用配备有二极管阵列检测器(dad)的agilent 1220infinity lc仪器进行hplc分析,配备有agilent eclipse plus c18反相色谱柱。使用merck的hplc级甲醇(有机流动相)。为了获得ph值为7.0的缓冲液(流动水相),在1000毫升去离子水中加入29.1毫升0.1m氢氧化钠和50毫升0.1m磷酸二氢钾(分析级)来制备溶液。缓冲液通过超声与真空过滤相结合的方法脱气。脱气过程中使用0.45μm whatmann滤纸。有机流动相在使用前也进行了脱气。使用等度溶剂系统实现色谱分离。hplc参数设置见下表。
[0059][0060]
在带有atr拟合的bruker alpha分光光度计上记录识别样品中官能团的ftir特征峰。在4000-400cm-1
范围内记录样品分析,峰值以波数(cm-1
)报告。对固体和液体样品进行了分析,没有进行任何改性。使用opus manager软件进行量化。
[0061]
在agilent 7820a仪器上使用hp-5色谱柱(30m
×
320μm
×
0.5μm)进行gc。采用火焰离子化检测器对样品进行检测。采用分流-无分流(split-split less)进样。下表提供了gc参数设置。
[0062]
为了确认合成化合物的纯度,使用stuart smp10数字仪器测定熔点。在分析之前,分析物样品必须在真空干燥器中干燥过夜。将少量粉末状分析物样品装入一端密封的玻璃毛细管中,深度约为4mm。将stuart装置设置在60℃平台温度。
[0063][0064]
合成步骤1:异烟酰胺13的制备
[0065][0066]
方案1:化合物13的制备
[0067]
方案1显示了该方法的第一合成步骤,其中在氢氧化钠水溶液的存在下将4-氰基吡啶12转化为异烟酰胺13。在考虑在反应的第二步中引入水合肼之前,该方法的第一步已充分完成(developed)。
[0068]
这些反应使用图3所示的微反应器装置进行。该微反应器装置由chemyx fusion 200模块化注射泵、10毫升sge玻璃注射器和硼硅酸盐玻璃板反应器ltf-v(体积:1.7毫升,通道尺寸:1毫米,几何形状:115
×
60
×
6毫米)和ltf-vs(体积:1.1毫升,通道尺寸:1毫米,几何形状:15
×
60
×
6毫米)构成。sge玻璃注射器通过内径为0.5mm的聚四氟乙烯管连接到一个微反应器板。使用放置在微反应器前的三通t-型连接器(omnifit labware,孔径:内径8.0mm,外径0.5-4mm)混合从两个注射器中泵出的试剂。为了让反应物溶液单向流动,两个反应物流中的每一个都串联了止回阀(cv出口:3302,1/4-28m至1/4-28f和cv入口:3301,1/4-28m至1/4-28f,15psi),如图3所示。
[0069]
将微反应器浸入温度控制油浴中,并通过一个装有两个注射器的chemyx fusion 200模块化注射泵将反应物输送到板上。10psi背压调节器(bpr)串联安装在反应器出口和收集瓶之间。下表1总结的初步实验表明,微反应器给出相当的性能。
[0070]
表1:ltf-v和ltf-vs反应器板初步试验的条件和结果总结。
[0071]
反应器[12](m)[naoh]t(℃)r
t
(分钟)12的转化率(%)选择性(%)ltf-v0.50.0590158593ltf-vs0.50.0590158189
[0072]
反应溶剂
[0073]
试剂和溶剂的相容性在流动系统中尤其重要,以确保产物和中间体保持在溶液中。不正确的溶剂和其他反应条件会导致试剂沉淀和压力升高。所研究的两种溶剂体系是水和水与甲醇的混合物。尽管在这些实验中使用了甲醇,但本领域技术人员会意识到类似的低级醇,例如乙醇,同样适用于作为反应溶剂。简要地同时考虑了反应温度的影响。
[0074]
在第一个初步实验中,使用两个注射器将4-氰基吡啶(0.5m)水溶液和氢氧化钠(0.05m)水溶液加料到微反应器中。在第二个初步实验中,使用两个注射器将4-氰基吡啶(0.5m)于7:3甲醇/水混合物中的溶液和氢氧化钠(0.05m)加料到微反应器中。反应分别在40℃和90℃的温度下进行,停留时间为15分钟。收集并使用离线hplc和gc分析样品。
[0075]
据观察,使用水仅导致反应器通道中的固体沉淀,从而降低了向所需中间体异烟酰胺13的转化率。在连续流系统中形成沉淀物是非常不可取的,因为它会导致连接器堵塞,最终导致系统内压力增加。因此,为反应选择合适的溶剂,使产物和中间体保持在溶液中是很重要的。
[0076]
与水混溶的溶剂甲醇的存在有助于溶解4-氰基吡啶、和由此产生的反应产物和副产物,并且未观察到反应通道和连接器内的沉淀。下表2总结了这些实验的结果,结果表明,使用醇(如甲醇)和升高温度促进了腈水解反应。
[0077]
表2:水和meoh:水混合物实验的条件和结果总结,其中[12]是4-氰基吡啶的浓度。
[0078]
运行[12](m)[naoh]溶剂系统t(℃)r
t
(分钟)转化率(%)10.50.05水40153520.50.05水90156230.50.05meoh/水(7:3)40154840.50.05meoh/水(7:3)901583
[0079]
试剂摩尔当量对异烟酰胺13转化率的影响
[0080]
反应中碱与腈的摩尔当量对于实现反应控制至关重要,尤其是在反应选择性方面。使用以下程序评估碱浓度对异烟酰胺13制备的影响。
[0081]
从第一个注射器(a)中加入4-氰基吡啶12于甲醇/水(7:3)溶液中的标准溶液(0.50m),从第二个注射器(b)中加入氢氧化钠于去离子水中的溶液(0.04m)。为了保持恒定的反应温度(90℃),将微反应器浸入温控油浴中。在停留15分钟后,将反应产物收集在小瓶中,以在进一步研究之前获得反应时间指示。为了研究试剂(4-氰基吡啶12和氢氧化钠)的相对化学计量比对产物13转化的影响,使用浓度变化范围为0至0.75m的不同氢氧化钠溶液重复所述实验。将每个流动反应的产物收集在小瓶中,并使用离线hplc进行分析。下表3示出了使用的反应参数和获得的结果的总结(见图4)。
[0082]
表3:用于研究摩尔当量对12转化为异烟酰胺13的影响的反应参数,其中[12]是4-氰基吡啶的浓度。
[0083][0084]
反应样品的分析提供了关于反应是否完成以及可能的副产物的初始信息。在此优化实验中分析样品期间,在hplc色谱图上观察到三个显著的峰。第一个峰对应于腈底物12,另一个峰对应于酰胺产物13,另一个峰对应于副产物异烟酸15。因此直接测量底物转化率,并根据该比率计算反应选择性。酰胺产物13的转化率用于确定最佳反应条件。
[0085]
图6显示了转化为所需异烟酰胺13和转化为副产物化合物异烟酸15之间的比较。
[0086]
从图4可以看出,在没有任何碱的情况下,转化率为5%。由于腈基不是具有特别的反应性,因此需要添加碱性催化剂来提高4-氰基吡啶12的反应性。然而,该反应所需的量以在连续流系统中产生所需的产率和选择性本身是未知的。很明显,随着化合物12:氢氧化钠的摩尔比增加,化合物12的转化率增加。当使用至少0.134m(0.2摩尔当量)氢氧化钠时,获得了100%的最大转化率。
[0087]
然而,即使当结果显示化合物12的转化率为100%时,增加氢氧化钠的浓度也会对酰胺13的选择性产生不利影响。优选地,4-氰基吡啶12与氢氧化钠的摩尔比在约1:0.1至约1:0.7,包括落入该范围内的任何特定比率,例如约1:0.15、约1:0.2、约1:0.25、约1:0.3、约1:0.35、约1:0.4、约1:0.45、约1:0.5、约1:0.55、约1:0.6、约1:0.65和约0.7。优选地,4-氰基吡啶12与氢氧化钠的摩尔比在约1:0.15至约1:0.6或约1:0.15至约1:0.4的范围内。
[0088]
停留时间对异烟酰胺13转化率的影响
[0089]
从第一个注射器(a)中添加4-氰基吡啶12于甲醇/水(7:3)溶液中的溶液(0.50m),并从第二个注射器(b)中泵送氢氧化钠于去离子水中的溶液(0.10m,0.2摩尔当量)。在不同的停留时间将反应产物收集在小瓶中。反应在90℃的恒温下进行。在八种不同的流速下研究了停留时间对转化为异烟酰胺13的影响,停留时间范围为1.5
–
32分钟。
[0090]
表4:用于研究停留时间对4-氰基吡啶12转化率影响的反应参数。
[0091][0092]
从上面表4中的结果和图6中的图表可以看出,12的转化率随着停留时间的增加而增加,并且从约7分钟的停留时间达到理想的转化率。优选地,12至13转化的停留时间为约7分钟至约32分钟,包括该范围内的任何持续时间。例如,停留时间可为约7分钟、约8分钟、约9分钟、约10分钟、约11分钟、约12分钟、约13分钟、约14分钟、约15分钟、约16分钟、约17分钟、约18分钟、约19分钟、约20分钟、约21分钟、约22分钟、约23分钟、约24分钟、约25分钟、约26分钟、约27分钟、约28分钟、约29分钟、约30分钟、约31分钟、以及约32分钟。优选地,用于转化12的停留时间为约7分钟至约20分钟、约7至约15分钟、或约10至约15分钟。
[0093]
反应温度对异烟酰胺13转化率的影响
[0094]
采用以下实验程序研究了反应温度对制备13的影响。将4-氰基吡啶12(0.50m)在meoh/h2o(7:3)溶液中的标准溶液放置在第一注射器(a)中,将氢氧化钠(0.10m)在去离子水中的溶液放置在第二注射器(b)中。在80℃和110℃之间的温度下,研究了温度对产物选择性和转化率的影响。微反应器中溶液的温度由恒温浴控制。将每个流动反应的产物收集在小瓶中,并立即使用离线hplc进行分析。为了确保获得的结果是由于微通道内发生的反应所导致,通过将装有样品的密封小瓶浸入室温下的烧杯水中,立即冷却收集的样品。下表5总结了实验中使用的不同参数。
[0095]
表5:温度对12至13转化率的影响。
[0096][0097][0098]
随着温度的升高,12的转化率逐渐增加,在95℃后达到100%转化率。从图5可以看出,结果表明13的转化率受反应温度的影响。随着温度从80-95℃逐渐升高,酰胺产品的转化率也提高到一定水平(约95%),之后随着副产物15的产生越来越多,转化率开始下降。因此不希望受任何特定理论的束缚,据信随着温度继续升高,反应有利于酸的形成,从而不会
提高酰胺的转化率。优选地,化合物12转化为化合物13的反应温度为约90至约105℃,更优选约90至约100℃。
[0099]
根据本研究中获得的结果,使用95℃的温度,异烟酰胺的转化率最佳。该温度使12的转化率达到99%,而形成较少的副产物(4%),导致异烟酰胺13达到96%的最高选择性。所有后续反应均采用以下最佳条件进行:停留时间10分钟,温度95℃。
[0100]
4-氰基吡啶12的浓度对异烟酰胺13转化率的影响
[0101]
在0.08m
–
1m范围内研究了化合物12浓度的影响。改变12的浓度,同时保持12和naoh之间的相对摩尔当量不变。使用以下实验程序研究浓度的影响。
[0102]
使用两个sge玻璃注射器和ptfe管(内径0.5mm),将4-氰基吡啶(0.08m在meoh/h2o 7:3中)和氢氧化钠(0.016m)进料到ltf-v微反应器。停留10分钟后,收集并分析样本。使用不同浓度的4-氰基吡啶12和氢氧化钠重复反应,如下表6所示。化合物12和氢氧化钠之间的摩尔当量保持恒定在1:0.2。使用离线hplc测定总转化率和选择性。
[0103]
表6:用于研究化合物12浓度对13转化率影响的反应参数。
[0104][0105][0106]
从上面表6所示的结果可以看出,4-氰基吡啶12的转化率受试剂12浓度的影响。一般来说,4-氰基吡啶浓度的增加导致生成的产物量增加。然而,令人惊讶的是,对于更高浓度,没有遵循同样的趋势。将12的浓度增加到1m导致转化率显著降低(5%)。
[0107]
合成步骤2:异烟酰肼(异烟肼)1的制备
[0108][0109]
方案2:化合物1的制备
[0110]
方案2显示了将中间体异烟酰胺13转化为异烟酰肼(异烟肼)1的反应步骤。如图2所示,通过两步合成路线研究了产物1的制备,其中对第二合成步骤,即上文所示的中间体化合物13与肼的反应,用原位制备的化合物13连续进行。
[0111]
该连续流反应装置由两个chemyx fusion 200模块化注射泵、三个10ml sge玻璃注射器(前两个注射器由一个泵控制,一个注射器由另一个泵控制)和不同的little things factory(ltf)玻璃反应器板(配有0.5mm id的ptfe管)构成。这些反应器板包括:1个ltf-v停留板(反应器体积:1.7ml,通道尺寸:1mm,几何尺寸:115
×
60
×
6mm),1个ltf-ms微混合器(反应器体积:0.2ml,通道尺寸:1mm,几何尺寸:115
×
60
×
6mm)和两个ltf-vs停留板(反应器体积:1.1ml,通道尺寸:1mm,几何尺寸:115
×
60
×
6mm)。该系统还包括单向流止回阀(cv出口:3302,1/4-28m至1/4-28f和cv入口:3301,1/4-28m至1/4-28f)和一个35psi背压调节器。这些反应器板布置在两个温度油浴中,其中第一油浴包含两个板:单个ltf-v停留板(用于合成酰胺13)和单个ltf-ms混合板,其包含两个输入通道(一个来自ltf-v停留板连续流,第二个作为水合肼入口进入流动物流)。第二油浴包含两个ltf-vs停留板(用于异烟酰肼1的合成)。使用ltf-ms微混合器将水合肼引入流动反应。反应的第二步需要充分混合,因此使用ltf-vs板。在第二步中使用两个停留板,只是为了增加停留时间。
[0112]
停留时间对异烟酰肼1转化率的影响
[0113]
使用图2所示的实验装置,从第一个注射器(a)中加入4-氰基吡啶12标准溶液(0.67m,在meoh/h2o 7:3中),从第二个注射器(b)中加入氢氧化钠溶液(0.134m,0.2当量),从第三个注射器(c)中加入水合肼溶液(1.34m,2.0当量)。使用10分钟的停留时间和95℃的浴温合成原位形成的中间体13。
[0114]
为了研究停留时间(在微反应器装置的第二部分中)对产物1转化率的影响,研究了添加水合肼后一系列的流速。将水合肼泵入系统,流速范围0.05
–
0.3ml/min的。这导致在该特定反应器装置中,肼反应的停留时间为2
–
25.2分钟。肼反应的温度保持恒定在110℃。将产物收集在小瓶中,并使用hplc进行分析。下表7示出了优化实验中使用的反应参数。
[0115]
表7:用于研究肼反应停留时间对1转化率影响的反应参数。
[0116][0117][0118]
一般来说,随着停留时间的增加,异烟酰肼1的转化率逐渐增加。上表7所示的停留时间(r
t
)是转化为中间体13的反应和转化为产物1的后续肼反应的合并停留时间。
[0119]
因此,从表7和图7的相应图表中可以看出,11分钟的有效停留时间(即21分钟的总时间减去转化为13的第一反应所用的10分钟)提供了95.5%的转化率,在19.1和25.2分钟的较高停留时间下,转化率略有提高。注意到酰胺中间体13到产物1具有完全选择性。因此,肼反应的停留时间在约10至约25分钟的范围内,包括该范围内的所有持续时间,例如10、11、12、13、14、15、16、17、18、19、20、21、22、23、24和25分钟。优选地,肼反应的停留时间在约
10到约20分钟的范围内。
[0120]
水合肼浓度对异烟酰肼1转化率的影响
[0121]
研究了相对于起始化合物12在0.67m(1当量)至1.675m(2.5当量)范围内的不同浓度的水合肼。肼反应的停留时间为11分钟(总停留时间为21分钟),而第一和第二反应的温度分别保持在95℃和110℃。使用4-氰基吡啶12(0.67m)、氢氧化钠(0.134m)和不同浓度的水合肼溶液。反应条件总结在下表8中。将样品收集在小瓶中,并使用hplc分析1的转化率和选择性。
[0122]
表8:用于研究水合肼浓度对产物1转化率影响的反应参数,其中r
t
是总停留时间。
[0123][0124][0125]
上面表8所示的结果表明,水合肼浓度对中间体13转化为异烟肼1有影响。相对于起始原料4-氰基吡啶12,水合肼的用量在1到2.5倍之间。结果表明,使用基于起始原料过量(2.0当量)的水合肼是非常有利的。优选地,4-氰基吡啶与水合肼的摩尔比在约1:1.50至约1:2.50的范围内,更优选在约1:1.75至约1:2.25的范围内,包括这些范围内包含的所有子范围。
[0126]
温度对异烟酰肼1转化率的影响
[0127]
将4-氰基吡啶(0.67m)加入注射器a中,将氢氧化钠(0.134m)从注射器b中添加,并将水合肼(1.34m)从注射器c中添加到连续流系统中。第一油浴(中间体13的合成)的温度保持在95℃,而第二个油浴的温度在90℃至120℃之间变化。为确保获得的结果是由于微通道内发生的反应所导致,立即冷却收集的样品,然后进行hplc分析。反应参数总结在下表9中。
[0128]
表9:用于研究肼反应温度对1转化率影响的反应参数。
[0129]
[0130]
肼反应中的温度对异烟酰肼1转化率的影响如上表结果所示。反应温度似乎对1转化率有显著影响。随着温度升高,产物1的生成量也呈非线性增加。
[0131]
以上对本发明的一些示例性实施方案的描述是为了说明如何制造和实施本发明。本领域的普通技术人员将知道,可以修改各种细节,从而获得进一步的实施方案,但是这些实施方案中的许多仍将保持在本发明的范围内。
[0132]
本说明书和附图中使用的不符合公制的单位,可借助以下换算系数换算为公制:
[0133]
1psi=6,895x103帕。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
