1.本发明涉及一种核酸复合体及包含该核酸复合体的医药组合物等。
背景技术:
2.作为核酸医药,已知有反义核酸、诱饵核酸、核酶、适体、sirna(small interfering rna,小干扰rna)、mirna(microrna,微小rna)、信使rna(mrna)等。其中,作用于mrna的核酸医药因其可控制细胞内的所有基因的通用性高,而期待将其临床应用于目前难以治疗的疾病中。即,核酸医药有望成为继低分子、抗体医药之后的下一代医药。然而,作为核酸医药的问题点,可列举:因核酸医药的相对较大的尺寸、磷酸骨架的负电荷引起的显著较低的细胞膜通透性、血中核酸酶引起的sirna分解等导致难以向核酸医药的作用点即细胞内传递(非专利文献1)。
3.针对该问题点,采用使传递手段适应核酸医药而解决的方法。作为传递手段之一,报告有靶配体与核酸的核酸复合体(复合核酸)。作为靶配体,可列举与在细胞表面表达的受体结合的形态。例如,作为相对于在肝实质细胞中高度表达的去唾液酸糖蛋白受体(asgpr)的配体,报告有多种利用n-乙酰基-d-半乳糖胺(galnac)等的核酸复合体(专利文献1、专利文献2、专利文献3、非专利文献2)。进而,作为对在巨噬细胞或树突细胞等免疫细胞中高度表达的甘露糖受体(cd206)传递的药物载体,报告有甘露糖偶联物及经甘露糖基化的核酸复合体(专利文献4)。又,报告有将胆固醇及脂肪酸等脂溶性化合物修饰于核酸医药,提高与血浆中的脂蛋白等的亲和性,由此实现向表达相对应的脂蛋白受体(ldl(low density lipoprotein,低密度脂蛋白)受体、hdl(high density lipoprotein,高密度脂蛋白)受体、清道夫受体srb1)的细胞的传递(非专利文献3)。现有技术文献专利文献
4.专利文献1:国际公开第2009/073809号专利文献2:国际公开第2014/179620号专利文献3:国际公开第2016/100401号专利文献4:国际公开第2018/004004号非专利文献
5.非专利文献1:nature reviews drug discovery.8,129-138(2009).非专利文献2:j.am.chem.soc.2014,136,16958-16961.非专利文献3:nucleic acids research,2019,vol.47,no.3,1082-1096.
技术实现要素:
[发明所要解决的问题]
[0006]
迄今为止提出多种靶配体、以及包含配体的核酸复合体,但现状下尚未发现可充分满足与受体的特异结合性的改善、核酸医药的细胞内吸收的增强者。
[解决问题的技术手段]
[0007]
本发明人等为了解决上述课题而进行了锐意研究,结果发现细胞选择性地吸收的核酸复合体。即,本发明涉及下述的[1]至[27]。[1]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(i)表示:[x为ch2或o;y为含有甘露糖或galnac的糖配体;n表示1至8的整数;z为含有寡核苷酸的基团]。[2]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(ii)表示:[y为含有甘露糖或galnac的糖配体;z为含有寡核苷酸的基团]。[3]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(iii)表示:[z为含有寡核苷酸的基团]。[4]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(iv)表示:
[z为含有寡核苷酸的基团]。[5]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(v)表示:[z为含有寡核苷酸的基团]。[6]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(vi)表示:[z为含有寡核苷酸的基团]。[7]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(vii)表示:
[z为含有寡核苷酸的基团]。[8]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(viii)表示:[z为含有寡核苷酸的基团]。[9]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(ix)表示:
[z为含有寡核苷酸的基团]。[10]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(x)表示:[z为含有寡核苷酸的基团]。[11]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(xi)表示:
[z为含有寡核苷酸的基团]。[12]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(xii)表示:[z为含有寡核苷酸的基团]。[13]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(xiii)表示:
[z为含有寡核苷酸的基团]。[14]一种核酸复合体或其药剂学上可接受的盐,该核酸复合体是以下述式(xiv)表示:[z为含有寡核苷酸的基团]。[15]如上述[1]至[14]中任一项所记载的核酸复合体,其中寡核苷酸为单链。[16]如上述[15]的核酸复合体,其中寡核苷酸经由3'端而结合。[17]如上述[15]的核酸复合体,其中寡核苷酸经由5'端而结合。[18]如上述[1]至[14]中任一项所记载的核酸复合体,其中寡核苷酸为双链。[19]如上述[18]的核酸复合体,其中寡核苷酸经由一条链的3'端而结合。[20]如上述[18]的核酸复合体,其中寡核苷酸经由一条链的5'端而结合。[21]一种医药组合物,其包含如上述[1]至[20]中任一项所记载的核酸复合体。
[22]如上述[21]所记载的医药组合物,其中对细胞中的靶基因的表达进行调节。[23]如上述[22]所记载的医药组合物,其中细胞为树突细胞、巨噬细胞或肝实质细胞。[24]如上述[1]至[20]中任一项所记载的核酸复合体,其用于对细胞中的靶基因的表达进行调节的方法。[25]如上述[24]所记载的核酸复合体,其中细胞为树突细胞、巨噬细胞或肝实质细胞。[26]一种调节对象细胞中的靶基因的表达的方法,其包括将如上述[1]至[20]中任一项所记载的核酸复合体或上述[21]的医药组合物施用至对象。[27]如上述[26]所记载的方法,其中细胞为树突细胞、巨噬细胞或肝实质细胞。[发明的效果]
[0008]
如下述药理试验中所示,本发明的核酸复合体被选择性地吸收至甘露糖受体或asgpr表达的细胞。通过将包含本发明的核酸复合体的医药组合物施用至哺乳动物(包括人类),而有可治愈各种关联疾病的可能性。
具体实施方式
[0009]
以下,对本发明的内容进行详细说明。
[0010]
关于本说明书中的化合物,存在为了方便起见其结构式表示某种异构物的情况,但不限定于为了方便的式子的记载,还包括化合物的结构上产生的所有几何异构物、光学异构物、旋转异构物、立体异构物、互变异构物等异构物以及异构物混合物,并且任一异构物均可为以任意比率包含各异构物的混合物。因此,例如本说明书中的化合物可能存在光学异构物及外消旋体,在本说明书中不限定于任一者,可为外消旋体,也可为各光学活性体的任一者,也可为以任意比率包含各光学活性体的混合物。
[0011]
又,本说明书中的化合物也有存在多晶型的情况,但同样不限定于任一者,可为任一晶型的单一物,也可为混合物,又,本说明书中的化合物也包括非晶体,并且本说明书中的化合物包含无水物与溶剂合物(尤其是水合物)。
[0012]
本说明书中的化合物也包含化合物的经同位素标记的化合物。经同位素标记的化合物除了1个或1个以上的原子经具有与自然界通常发现的原子质量或质量数不同的原子质量或质量数的原子取代以外,与化合物相同。可掺入本说明书中的化合物的同位素例如为氢、碳、氮、氧、氟、磷、硫、碘、及氯的同位素,包括2h、3h、
11
c、
14
c、
15
n、
18
o、
18
f及
35
s等。
[0013]
本说明书中的所谓“药剂学上可接受的盐”只要为与化合物形成盐者,则无特别限定,具体而言,例如可列举:酸加成盐、金属盐、铵盐、有机胺加成盐、氨基酸加成盐等盐。
[0014]
作为酸加成盐的优选例,例如可列举:盐酸盐、氢溴酸盐、硫酸盐、硝酸盐、磷酸盐等无机酸盐;或乙酸盐、琥珀酸盐、富马酸盐、马来酸盐、酒石酸盐、柠檬酸盐、乳酸盐、硬脂酸盐、苯甲酸盐、甲磺酸盐、对甲苯磺酸盐、苯磺酸盐等有机酸盐。作为金属盐的优选例,例如可列举:钠盐、钾盐等碱金属盐;镁盐、钙盐等碱土金属盐;铝盐、锌盐等。作为铵盐的优选例,例如可列举:铵、四甲基铵等的盐。作为有机胺加成盐的优选例,例如可列举:吗啉、哌啶等的加成盐。
作为氨基酸加成盐的优选例,例如可列举:赖氨酸、甘氨酸、苯丙氨酸、天冬氨酸、谷氨酸等的加成盐。
[0015]
于以游离体获得本说明书中的化合物的情形时,可依照常规方法转换为化合物可形成的盐或该等的水合物的状态。
[0016]
于以盐或水合物的形式获得本说明书中的化合物的情形时,可依照常规方法转换为游离体。
[0017]
又,关于本说明书中的化合物而获得的各种异构物(例如几何异构物、光学异构物、旋转异构物、立体异构物、互变异构物等)可通过使用通常的分离方法,例如再结晶、非镜像异构物盐法、酶切法、各种色谱法(例如薄层色谱法、管柱色谱法、气相色谱法、高效液相色谱法等)进行纯化而单离。
[0018]
本发明的医药组合物可通过将药剂学上可接受的添加物与核酸复合体或其药剂学上可接受的盐混合而制造。本发明的医药组合物例如可依照日本药典第十七修订版的制剂总则中所记载的方法等已知的方法而制造。
[0019]
本发明的医药组合物可根据其剂型而适当地施用至患者。
[0020]
本发明的核酸复合体的施用量依症状的程度、年龄、性别、体重、施用形态或盐的种类、疾病的具体的种类等而异,通常在成人的情形时,以换算为寡核苷酸的施用量计,经口施用时每天将约30μg~10g、优选为100μg~1g,注射施用时每天将约1μg~1g、优选为100μg~300mg分别1次施用或分为多次施用。
[0021]
本发明的核酸复合体为糖配体经由接头与寡核苷酸结合而成的核酸复合体,糖配体含有o、n、或c结合、优选为o结合甘露糖或galnac。即,本发明的核酸复合体具有(糖配体)-(接头)-(寡核苷酸)的结构。
[0022]
在一实施方式中,核酸复合体含有2个以上糖、优选为3个或4个糖。在一实施方式中,核酸复合体含有至少3个甘露糖或至少3个galnac,该等核酸复合体可分别以巨噬细胞或肝实质细胞为靶而传递。
[0023]
<关于糖配体>甘露糖受体在cd206高度表达的细胞、例如巨噬细胞或树突细胞等特定的细胞中高度表达。甘露糖偶联物及经甘露糖基化的药物载体对cd206表达出较高的结合亲和性,并已成功地用于将药物分子、例如寡核苷酸传递到巨噬细胞或树突细胞等细胞中。
[0024]
asgpr在肝实质细胞等特定的细胞中高度表达。galnac偶联物及经galnac化的药物载体对asgpr表达出较高的结合亲和性,并已成功地用于将药物分子、例如寡核苷酸传递到肝实质细胞等细胞中。
[0025]
糖配体意指源自可与在靶细胞中表达的受体结合的糖类的基团,作为本发明的核酸复合体中优选的糖配体的方面之一,可例示以下结构。波形线意指与接头的结合键。
[0026]
<关于寡核苷酸>作为本发明的核酸复合体中的寡核苷酸,可使用已知用作核酸医药的寡核苷酸。在本说明书中,核酸医药意指以反义核酸、诱饵核酸、核酶、sirna、mirna、antimirna及mrna等形式使用的核苷酸。
[0027]
又,寡核苷酸也可为单链或双链的寡核苷酸。
[0028]
本发明的核酸复合体中的接头与寡核苷酸可在寡核苷酸等的核苷酸处结合,例如在寡核苷酸的3'端或5'端结合。在寡核苷酸为双链的情形时,接头优选为与构成双链核酸的正义链的3'端或5'端结合,但不限定于该结合。
[0029]
核酸复合体中的寡核苷酸所结合的接头的数量不限于1个,也可为2个以上。
[0030]
在若干实施方式中,例如为了提高稳定性,可在突出端包含特定的碱基,或者在单链突出端(例如,5'突出端或3'突出端、或该两者)包含修饰核苷酸或核苷酸代用物。
[0031]
构成本发明的核酸复合体的寡核苷酸只要在导入哺乳动物细胞的情形时具有控制靶基因的表达的能力,则可为任意形状,可适宜地使用单链的寡核苷酸或双链的寡核苷酸。
[0032]
作为寡核苷酸,只要为核苷酸或具有与核苷酸同等功能的分子的聚合物,则可为任意分子,例如可列举:作为脱氧核糖核苷酸的聚合物的dna、作为核糖核苷酸的聚合物的rna、作为dna与rna的聚合物的嵌合核酸。又,在dna、rna及嵌合核酸中,也可为至少一个脱氧核糖核苷酸或核糖核苷酸等核苷酸被具有与核苷酸同等功能的分子取代而成的核苷酸聚合物。再者,rna中的尿嘧啶(u)在dna中可单义地改称为胸腺嘧啶(t)。
[0033]
作为具有与核苷酸同等功能的分子,例如可列举对核苷酸施加修饰而成的核苷酸衍生物等,例如为了与dna或rna相比而提高或稳定核酸酶耐性,提高与互补链核酸的亲和性,提高细胞通透性,或使其可视化,可适当使用对脱氧核糖核苷酸或核糖核苷酸施加修饰而成的分子等。
[0034]
作为核苷酸衍生物,例如可列举糖部修饰核苷酸、磷酸二酯键修饰核苷酸、碱基修
饰核苷酸等糖部、磷酸二酯键及碱基的至少一者被修饰的核苷酸等。
[0035]
作为糖部修饰核苷酸,只要为对于核苷酸的糖的化学结构的一部分或全部,以任意取代基修饰或取代而成者、或以任意原子取代而成者,则可为任意者,可优选使用2'-修饰核苷酸。
[0036]
作为2'-修饰核苷酸,例如可列举核糖的2'-oh基被选自由or、r、r'or、sh、sr、nh2、nhr、nr2、n3、cn、f、cl、br及i所组成的组(r为烷基或芳基,优选为碳数1~6的烷基,r'为亚烷基,优选为碳数1~6的亚烷基)的取代基取代的2'-修饰核苷酸,作为2'-修饰,优选可列举以f、甲氧基及乙氧基进行的取代。又,也可为通过亚甲基桥接核糖的2'位的氧原子与4'位的碳的2'-修饰核苷酸、即锁核酸。
[0037]
作为磷酸二酯键修饰核苷酸,只要为对于核苷酸的磷酸二酯键的化学结构的一部分或全部,以任意取代基修饰或取代而成者、或以任意原子取代而成者,则可为任意者,例如可列举:磷酸二酯键被硫代磷酸酯键取代的核苷酸、磷酸二酯键被二硫代磷酸酯键取代的核苷酸、磷酸二酯键被膦酸烷基酯键取代的核苷酸、磷酸二酯键被胺基磷酸酯键取代的核苷酸等,优选可列举磷酸二酯键被硫代磷酸酯键取代的核苷酸。
[0038]
寡核苷酸也包含其分子中的一部分或全部原子被质量数不同的原子(同位素)取代而成者。
[0039]
在一方面中,本发明的核酸复合体中的寡核苷酸对细胞中的靶基因的表达进行调节。
[0040]
在一方面中,本发明的核酸复合体中的寡核苷酸经由接头与配体(也称为接头配体)结合。
[0041]
<关于接头>作为本发明中的“接头”,可使用可用作核酸复合体的任意接头。
[0042]
作为接头结构的例示,例如可采用国际公开第2009/073809号、国际公开第2013/075035号、国际公开第2015/105083号所公开的结构。
[0043]
在一实施方式中,接头包含至少1个可切断的结合基。
[0044]
所谓可切断的结合基是在细胞外十分稳定,但若进入靶细胞内则被切断而释放出与接头结合的部分的基团。
[0045]
例如,基于磷酸的可切断的结合基可被将磷酸基分解或水解的药剂所切断。在细胞内切断磷酸基的例子为磷酸酶等酶。基于磷酸的结合基的优选的实施方式为-o-p(o)(oh)-o-或o-p(s)(oh)-o-。
[0046]
作为接头的实施方式,为下述结构的任一者,x、y分别独立存在,x表示ch2或o,y表示糖配体,z表示含有寡核苷酸的基团,n表示1至8的整数。
实施例
[0047]
本发明的核酸复合体例如可通过以下的制造例所记载的方法而制造,又,该化合物的效果可通过以下的试验例所记载的方法进行确认。但该等为例示者,本发明在任意情形时均不限制于以下具体例,又,可在不脱离本发明的范围的范围内变化。本发明中的实施例可使用本领域技术人员已知的合成化学技术进行制造。
[0048]
又,本说明书所使用的缩写是本领域技术人员所周知的惯用的缩写。本说明书中使用以下缩写。cbz:苄氧羰基ctc:2-氯三苯甲基氯dce:1,2-二氯乙烷dcm:二氯甲烷dipea:n,n-二异丙基乙基胺dmf:n,n-二甲基甲酰胺dmso:二甲基亚砜edci:1-乙基-3-(3-二甲胺基丙基)碳二酰亚胺盐酸盐etoac:乙酸乙酯esi:电喷雾离子化fmoc:9-芴基甲氧基羰基hbtu:1-[双(二甲胺基)亚甲基]-1h-苯并三唑鎓3-氧化物六氟磷酸盐hobt:1-羟基苯并三唑ims:工业用改性醇maldi-tof-ms:基质辅助激光解吸电离-飞行时间型质谱仪meoh:甲醇mtbe:甲基叔丁基醚naome:甲醇钠tfa:三氟乙酸1h-nmr:质子核磁共振谱分析ms:质谱分析hplc:高效液相色谱法
[0049]
以下的实施例及制造例中的“室温”通常表示约10℃至约35℃。%只要无特别说明,则表示重量或体积百分比。
[0050]
质子核磁共振谱的化学位移以相对于四甲基硅烷的δ单位(ppm)记录,偶联常数以赫兹(hz)记录。模式为s:单峰、d:双峰、t:三重峰、q:四重峰、quin:五重峰、m:多重峰、br:宽峰、br.s:宽单峰。
[0051]
关于硅胶管柱色谱法,硅胶使用merck公司制造的silica gel60(70-230目、或230-400目astm)、fuji silysia chemical公司制造的psq60b,或者使用预装柱(管柱:yamazen公司制造的hi-flash
tm column(silicagel)、或biotage公司制造的biotage
tm
snap ultra silica cartridge)。
[0052]
化合物的命名除了通常所使用的试剂以外,使用以“e-notebook”第12或13版(perkinelmer公司)或“marvinsketch”第16版(chemaxon公司)所示的命名。
[0053]
以下述制造例为参考,可通过本领域技术人员所公知的方法进行制造。
[0054]
化合物6的合成流程
[0055]
化合物2的合成
[0056]
在乙酸肼(5.19g,56.4mmol)中添加dmf(200ml),在55℃搅拌后,将反应混合物冷却为15℃,添加1-o,2-o,3-o,4-o,6-o-五乙酰基-β-d-甘露糖(化合物1)(20.0g,51.2mmol),在15℃搅拌16小时。将反应混合物添加至水中,利用etoac进行萃取。利用饱和食盐水洗净合并的有机层,利用无水硫酸钠加以干燥,过滤后,在减压下进行浓缩而获得化合物2(14.3g)。1h-nmr(400mhz,cdcl3)δ(ppm):1.97-2.00(m,3h),2.03(s,3h),2.08(s,3h),2.14(s,3h),4.11-4.15(m,1h),4.16-4.32(m,3h),5.14-5.34(m,3h),5.37-5.45(m,1h)。
[0057]
化合物3的合成
[0058]
在dcm(50ml)中添加化合物2(5.00g,14.4mmol)、2,2,2-三氯乙腈(20.7g,143mmol)及cs2co3(5.14g,15.8mmol),在25℃搅拌2小时。对反应混合物进行过滤,将滤液在减压下进行浓缩。通过硅胶管柱色谱法(50:1-20:1石油醚/etoac)纯化残渣而获得化合物3(2.70g)。1h-nmr(400mhz,cdcl3)δ(ppm):2.01(s,3h),2.04(s,2h),2.08(d,j=6.8hz,6h),2.20(s,3h),4.15-4.22(m,2h),4.24-4.32(m,1h),5.37-5.42(m,2h),5.46-5.49(m,1h),6.28(d,j=1.8hz,1h),8.79(s,1h)。
[0059]
化合物4的合成
[0060]
将化合物3(14.0g,28.4mmol)、6-(cbz-胺基)-1-己醇(8.57g,34.1mmol)及分子筛4a(10.0g)添加至dcm(140ml)中,在0℃搅拌30分钟。继而,在-65℃将三甲基硅烷基三氟甲磺酸酯(6.32g,28.4mmol)滴加至反应混合物中,将反应混合物在25℃搅拌16小时。利用dcm稀释反应混合物,过滤后,利用饱和碳酸氢钠水溶液、水及食盐水洗净滤液,利用无水硫酸钠加以干燥,过滤后,在减压下进行浓缩。通过硅胶管柱色谱法(50:1-20:1石油醚/etoac)纯化残渣而获得化合物4(7.50g)。
[0061]
化合物5的合成
[0062]
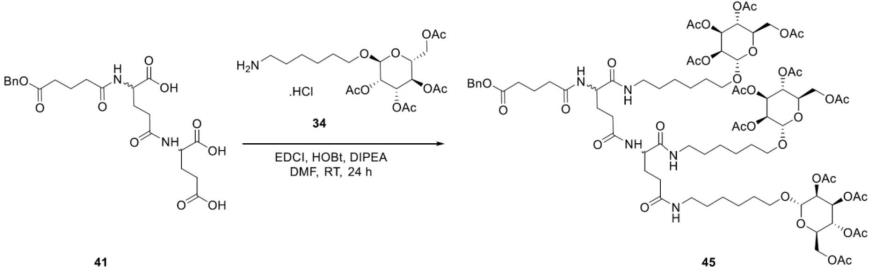
将化合物4(7.50g,12.9mmol)溶解于meoh(300ml)中,添加naome(2.79g,51.6mmol),在室温下反应3小时。将反应混合物的一部分浓缩,注入冷水中,添加乙酸直至ph=5为止,通过硅胶管柱色谱法(50:1-20:1dcm/meoh)纯化所获得的粗产物而获得化合物5(3.90g)。1h-nmr(400mhz,cdcl3)δ(ppm):1.26-1.63(m,8h),3.14(d,j=6.3hz,2h),3.34(d,j=8.8hz,1h),3.54-3.74(m,2h),3.80-4.01(m,4h),4.78(s,1h),5.01-5.12(m,2h),5.16-5.47(m,6h),7.28-7.41(m,5h)。
[0063]
化合物6的合成
[0064]
在干燥10%钯-碳(0.40g)中添加meoh(40ml)、化合物5(3.90g,9.43mmol),在25℃、氢气压力下(50psi)搅拌3小时。对反应混合物进行过滤,在减压下进行浓缩而获得化合
物6(1.50g)。1h-nmr(400mhz,meod)δ(ppm):1.38-1.49(m,4h),1.61(dquin,j=13.3,6.9,6.9,6.9,6.9hz,4h),2.73-2.88(m,2h),3.31(dt,j=3.3,1.6hz,1h),3.43(dt,j=9.7,6.1hz,1h),3.48-3.89(m,8h)。
[0065]
化合物8的合成
[0066]
在6-叠氮基己酸(化合物7)(50.2mg,319μmol)及化合物6(268mg,959μmol)的dmf(3ml)溶液中添加hobt(129mg,959μmol)、dipea(248mg,1.92mmol)及edci(183mg,959μmol),在25℃搅拌3小时。通过制备型hplc(tfa条件)纯化反应混合物而获得化合物8(20.1mg)。1h-nmr(400mhz,dmso-d6)δ(ppm):1.24-1.54(m,14h)2.04(t,j=7.40hz,2h)3.00(q,j=6.53hz,2h)3.23-3.40(m,12h)3.55-3.66(m,2h)4.57(s,1h)7.75(br s,1h)
[0067]
化合物11的合成流程
[0068]
化合物10的合成在包含ctc树脂(0.50g,0.50mmol,1.00mmol/g)与n-[(9h-芴-9-基甲氧基)羰基]-谷氨酸5-叔丁酯(化合物9)(212mg,0.50mmol)的混合物中添加dcm(30ml)及dipea(258mg,2.00mmol),将反应混合物搅拌2小时。在反应混合物中添加meoh(0.2ml),并搅拌30分钟。在反应混合物中添加6-叠氮基己酸(化合物7)(118mg,0.75mmol)、dipea(258mg,2.00mmol)、hbtu(270mg,712umol)及dmf(3ml),在25℃搅拌1小时。在反应混合物中添加90%tfa/10%dcm,并搅拌2小时。对反应混合物进行过滤,在减压下浓缩滤液而获得化合物10(110mg)。
[0069]
化合物11的合成
[0070]
在化合物10(50.0mg,174μmol)及化合物6(292mg,1.05mmol)的dmf(3ml)溶液中添加hobt(141mg,1.05mmol)、dipea(270mg,2.10mmol)及edci(200mg,1.05mmol)。将反应混合物在25℃搅拌16小时。通过制备型hplc(tfa条件)进行纯化而获得化合物11(18.1mg)。1h-nmr(400mhz,dmso-d6)δ(ppm):1.16-1.56(m,24h),1.62-1.73(m,1h),1.76-1.87(m,1h),2.00-2.07(m,2h),2.09-2.15(m,2h),2.95-3.07(m,4h),3.22-3.41(m,18h),3.65(br s,6h),4.10-4.19(m,1h),4.57(d,j=1.00hz,2h),7.74-7.93(m,3h)。
[0071]
化合物12的合成
[0072]
在ctc树脂(1.00g,1.00mmol,1.00mmol/g)及n-[(9h-芴-9-基甲氧基)羰基]-谷氨酸5-叔丁酯(化合物9)(511mg,1.20mmol)中添加dcm(20ml)、dipea(516mg,4.00mmol),对反应混合物通入氮气(n2)并搅拌2小时。在反应混合物中添加meoh(1ml),并搅拌30分钟。将20%哌啶的dmf(30ml)溶液添加至反应混合物中,并搅拌30分钟,利用dmf洗净树脂,添加n-fmoc-谷氨酸1-叔丁酯(851mg,2.00mmol)、hbtu(720mg,1.90mmol)及dipea(516mg,4.00mmol)的dmf(5ml)溶液,在25℃搅拌1小时。添加20%哌啶的dmf(30ml)溶液,搅拌30分钟,并利用dmf洗净树脂。继而,添加6-叠氮基己酸(化合物7)(0.30g,1.91mmol)、hbtu(720mg,1.90mmol)及dipea(516mg,4.00mmol)的dmf(5ml)溶液,在25℃搅拌1小时,利用dmf洗净树脂,并利用meoh进行洗净,在减压下使其干燥。在所获得的残渣(0.60g)中添加50%tfa/50%dcm,搅拌2小时,在减压下浓缩反应混合物而获得化合物12(240mg)。
[0073]
化合物13的合成化合物13的合成是依照化合物12的合成方法,使用叠氮-peg8-酸(azido-peg8-acid)(cas no.1214319-92-2)代替6-叠氮基己酸,由此获得化合物13(350mg)。1h-nmr(400mhz,cdcl3)δ(ppm):2.01(s,2h),2.18(s,2h),2.32-2.63(m,6h),3.40(t,j=5.0hz,2h),3.54-3.71(m,32h),4.49(s,2h),7.67-8.03(m,2h)。
[0074]
化合物14的合成
[0075]
在化合物12(0.20g,481μmol)及化合物7(1.08g,3.85mmol)的dmf(0.5ml)溶液中添加dipea(996mg,7.70mmol)、hobt(520mg,3.85mmol)及edci(738mg,3.85mmol)。将反应混合物在25℃搅拌16小时,通过制备型hplc(tfa条件)进行纯化而获得化合物14(103mg)。1h-nmr(400mhz,meod)δ(ppm):1.26-1.72(m,32h),1.82-1.97(m,2h),2.03-2.13(m,2h),2.22-2.41(m,6h),3.14-3.24(m,6h),3.37-3.47(m,3h),3.49-3.55(m,3h),3.60(t,j=9.5hz,3h),3.65-3.86(m,15h),4.22-4.42(m,2h),4.73(s,3h)。
[0076]
化合物15的合成
[0077]
在化合物13(0.20g,275μmol)及化合物7(616mg,2.20mmol)的dmf(0.5ml)溶液中添加dipea(570mg,4.41mmol)、hobt(298mg,2.20mmol)及edci(423mg,2.20mmol),将反应混合物在25℃搅拌16小时。通过制备型hplc(tfa条件)纯化反应混合物而获得化合物15(127mg)。1h-nmr(400mhz,meod)δ(ppm):1.23-1.68(m,24h),1.78-1.95(m,2h),2.04-2.19(m,2h),2.23-2.31(m,2h),2.33-2.42(m,2h),2.48-2.60(m,2h),3.10-3.26(m,6h),3.35-3.46(m,5h),3.48-3.56(m,3h),3.57-3.84(m,50h),4.23-4.41(m,2h),4.73(s,3h)。
[0078]
化合物17的合成流程
[0079]
化合物16的合成在ctc树脂(0.50mmol,0.50g,1.00mmol/g)及n-[(9h-芴-9-基甲氧基)羰基]-谷氨酸5-叔丁酯(化合物9)(212mg,0.50mmol)中添加dcm(30ml)及dipea(258mg,2.00mmol)。将反应混合物搅拌2小时后,添加meoh(0.2ml),并搅拌30分钟。在反应混合物中添加hbtu(360mg,950μmol)及dipea(258mg,2.00mmol)的dmf(3ml)溶液及n-fmoc-谷氨酸1-叔丁酯(425mg,1.00mmol),在25℃搅拌1小时后,添加n-fmoc-谷氨酸1-叔丁酯(425mg,1.00mmol)、hbtu(360mg,950μmol)及dipea(258mg,2.00mmol)的dmf(3ml)溶液,在25℃在1小时内添加。继而,添加6-叠氮基己酸(化合物7)(118mg,750μmol)、hbtu(360mg,950μmol)、dipea(258mg,2.00mmol)并搅拌1小时。添加90%tfa/10%dcm,在室温下搅拌2小时。对反应混合物进行过滤,在减压下进行浓缩而获得化合物16(200mg)。
[0080]
化合物17的合成
[0081]
在化合物16(50.0mg,91.8μmol)及化合物7(256mg,918μmol)的dmf(3ml)溶液中添加hobt(124mg,918μmol)、dipea(237mg,1.84mmol)及edci(176mg,918μmol),将反应混合物在25℃搅拌16小时。通过制备型hplc(tfa条件)纯化反应混合物而获得化合物17(18.2mg)。1h-nmr(400mhz,dmso-d6)δ(ppm):1.19-1.56(m,40h),1.66(br s,3h),1.83(br s,3h),2.00-2.15(m,7h),2.94-3.05(m,8h),3.29(br d,j=5.02hz,24h),3.52-3.71(m,
24h),4.13(br s,3h),4.57(s,4h),7.74-7.99(m,7h)。
[0082]
化合物21及22的合成流程
[0083]
化合物19的合成
[0084]
在半乳糖胺五乙酸酯化合物18(12.0g,30.8mmol)、6-(cbz-胺基)-1-己醇(10.3g,41.1mmol)及zncl2(5.60g,41.1mmol)(在使用前在110℃减压干燥1小时)的混合物中添加dce(120ml),将反应混合物在70℃搅拌3小时。利用etoac及碳酸氢钠水溶液稀释反应混合物,将混合物搅拌10分钟,进行硅藻土(注册商标)过滤,并利用etoac洗净。利用水及食盐水洗净滤液。利用etoac萃取水层,利用硫酸钠干燥有机层,过滤后,在减压下进行浓缩,通过硅胶管柱色谱法(2:1石油醚/etoac)纯化所获得的残渣而获得化合物19(14.0g)。1h-nmr(400mhz,cdcl3)δ(ppm):1.35(d,j=3.8hz,4h),1.45-1.66(m,4h),1.77(s,2h),1.94(s,3h),2.00(s,3h),2.05(s,3h),2.14(s,3h),3.20(qq,j=13.4,6.8hz,2h),3.48(dt,j=9.6,6.5hz,1h),3.79-4.03(m,3h),4.08-4.19(m,2h),4.65(d,j=8.3hz,1h),4.90(s,1h),5.04-5.17(m,2h),5.26(dd,j=11.2,3.1hz,1h),5.30(s,1h),5.34(d,j=2.8hz,1h),5.94(d,j=8.8hz,1h),7.28-7.42(m,5h)。
[0085]
化合物20的合成
[0086]
在化合物19(7.50g,12.9mmol)中添加meoh(300ml)及naome(2.79g,51.7mmol),在室温下搅拌3小时。将反应混合物的一部分浓缩,注入冷水中,添加乙酸直至ph=5为止。继而,通过硅胶管柱色谱法(50:0-50:1dcm/meoh)纯化所获得的粗产物而获得化合物20(4.00g)。1h-nmr(400mhz,meod)δ(ppm):1.19-1.35(m,4h),1.40-1.56(m,4h),1.93(s,2h),3.06(t,j=7.0hz,2h),3.27(dt,j=3.3,1.6hz,1h),3.31(s,1h),3.37-3.49(m,2h),3.55(dd,j=10.8,3.3hz,1h),3.65-3.76(m,2h),3.78-3.94(m,3h),4.31(d,j=8.5hz,1h),
5.02(s,2h),7.17-7.44(m,5h)。
[0087]
化合物21的合成
[0088]
在氩气环境下向干燥10%钯-碳(240mg)中添加meoh(25ml)及化合物20(2.40g,5.28mmol),在氢气压力下(50psi)在25℃搅拌3小时。对反应混合物进行过滤,在减压下浓缩滤液而获得化合物21(0.66g)。1h-nmr(400mhz,meod)δ(ppm):1.30-1.44(m,4h),1.52-1.66(m,4h),1.98(s,3h),2.76-2.90(m,2h),3.31(dt,j=3.3,1.6hz,2h),3.44-3.51(m,2h),3.58(dd,j=10.8,3.3hz,1h),3.73-3.78(m,2h),3.82-3.95(m,3h),4.34(d,j=8.5hz,1h)。
[0089]
化合物22的合成在化合物19(202mg,0.35mmol)中添加乙醇(6ml)、10%钯-碳(50%含水品)(42mg),在氢气环境下在室温下搅拌3小时。利用硅藻土(注册商标)过滤反应混合物,利用乙醇洗净。在减压下浓缩滤液而获得化合物22(159mg)。1h-nmr(600mhz,dmso-d6)δ(ppm):0.64(br s,1h)1.27(br s,5h)1.33-1.52(m,4h)1.67(br s,1h)1.80(s,3h)2.61(br t,j=6.88hz,2h)3.40-3.60(m,5h)3.58-3.77(m,4h)4.23(br d,j=8.25hz,1h)4.31-4.97(m,5h)7.53-7.71(m,1h);ms(esi ):m/z 321[m h] 。
[0090]
化合物23的合成
[0091]
在化合物12(602mg,1.88mmol)及化合物21(601mg,1.88mmol)的dmf(0.5ml)溶液中添加dipea(485mg,3.76mmol)、hobt(254mg,1.88mmol)及edci(360mg,1.88mmol),将反应混合物在25℃搅拌16小时。通过制备型hplc(tfa条件)纯化残渣而获得化合物23(104mg)。1h-nmr(400mhz,meod)δ(ppm):1.29-1.70(m,32h),1.82-2.01(m,11h),2.08(s,2h),2.22-2.38(m,8h),3.18(d,j=6.3hz,6h),3.42-3.52(m,6h),3.60(d,j=10.5hz,3h),
3.71-3.95(m,14h),4.28(s,1h),4.36(d,j=8.3hz,3h)。
[0092]
化合物24的合成以与化合物23的合成同样的方式,使用化合物16代替化合物12,由此获得化合物24。1h-nmr(600mhz,dmso-d6)δ(ppm):1.15-1.59(m,44h)1.68(br d,j=9.90hz,3h)1.79(s,12h)1.82-1.92(m,5h)1.97-2.22(m,9h)3.02(br d,j=5.50hz,10h)3.32-3.58(m,19h)3.61-3.72(m,13h)4.06-4.27(m,8h)4.43(br s,4h)4.46-4.59(m,9h)7.59(br d,j=8.80hz,4h)7.74(br s,1h)7.83(br s,3h)7.89(br s,3h);ms(esi ):m/z 1754[m h] 。
[0093]
化合物28的合成流程
[0094]
化合物25的合成
在包含ctc树脂(1.33mmol/g,0.375g,0.50mmol)与化合物9(212mg,0.50mmol)的混合物中添加dcm(30ml),并滴加dipea(0.35ml,2.00mmol),将反应液搅拌2小时。在反应液中添加meoh(0.2ml,4.94mmol),并搅拌35分钟。在反应液中添加20%哌啶的dmf(15ml)溶液并搅拌30分钟。对反应混合物进行过滤,利用dmf洗净所获得的固体。继而添加n-fmoc-d-谷氨酸1-叔丁酯(213mg,0.499mmol)及n-fmoc-l-谷氨酸1-叔丁酯(213mg,0.499mmol)、hbtu(360mg,0.949mmol)、dmf(3ml)及dipea(0.35ml,2.00mmol),对反应混合物通入氮气并搅拌1小时。添加20%哌啶的dmf(15ml)溶液,并搅拌30分钟。然后利用dmf洗净树脂。继而添加n-fmoc-d-谷氨酸1-叔丁酯(213mg,0.499mmol),n-fmoc-l-谷氨酸1-叔丁酯(213mg,0.499mmol)、hbtu(360mg,0.949mmol)及dmf(3ml),缓慢滴加dipea(0.35ml,2.00mmol)。对反应混合物通入氮气并搅拌80分钟。然后,利用dmf洗净树脂,并利用meoh进行洗净,在减压下使其干燥。在所获得的残渣中添加50%tfa/50%dcm,搅拌2小时并过滤后,在减压下浓缩滤液而获得化合物25(307.3mg)。ms(esi)m/z:610[m h]
1
h-nmr(600mhz,dmso-d6)δ(ppm):1.77(m,5h),1.94(m,3h),2.12-2.23(m,6h),2.26(m,2h),2.38(br t,j=7.34hz,2h),4.13-4.21(m,3h),5.09(s,2h),7.30-7.40(m,5h),8.05-8.15(m,3h)。
[0095]
化合物26的合成在化合物22(154mg,0.344mmol)与化合物25(21.0mg,0.034mmol)的dmf(2ml)溶液中添加hobt(52.8mg,0.344mmol)、dipea(0.12ml,0.689mmol)及edci(66.0mg,0.344mmol)。将反应混合物在室温下搅拌18小时,添加水及etoac、少量甲醇进行萃取,利用硫酸钠干燥
有机层。过滤后在减压下进行浓缩。通过制备型hplc纯化残渣而获得化合物26(16.7mg)。ms(esi)m/z:1162[m 2h]
1
h-nmr(600mhz,dmso-d6)δ(ppm):1.23(br s,16h),1.31-1.41(m,8h),1.41-1.48(m,8h),1.60-1.73(m,4h),1.77(s,12h),1.78-1.87(m,4h),1.89(s,12h),1.99(s,12h),2.01-2.07(m,2h),2.10(s,12h),2.11-2.21(m,6h),2.33-2.37(m,2h),2.95-3.07(m,8h),3.37-3.43(m,4h),3.65-3.72(m,4h),3.82-3.90(m,4h),3.98-4.05(m,12h),4.09-4.20(m,3h),4.48(d,j=8.44hz,4h),4.97(dd,j=11.19,3.12hz,4h),5.08(s,2h),5.21(d,j=3.30hz,4h),7.30-7.39(m,5h),7.80(m,8h),7.86-7.95(m,3h)。
[0096]
化合物27的合成在化合物26(19.0mg,8.18μmol)的乙醇(3ml)溶液中添加10%钯-碳(50%含水品)(5.3mg)。将反应混合物在氢气环境下在室温下搅拌3小时。利用硅藻土(注册商标)过滤反应混合物,利用乙醇洗净。在减压下浓缩滤液而获得化合物27(19.6mg)。1h-nmr(600mhz,dmso-d6)δ(ppm):1.24(br s,22h),1.30-1.40(m,8h),1.45(br s,8h),1.61-1.74(m,4h),1.77(s,12h),1.80-1.87(m,2h),1.89(s,12h),1.99(s,12h),2.01-2.07(m,2h),2.10(s,12h),2.12-2.25(m,4h),2.93-3.06(m,8h),3.36-3.46(m,4h),3.64-3.72(m,4h),3.82-3.91(m,4h),3.98-4.20(m,16h),4.50(br d,j=8.07hz,4h),4.94-5.02(m,4h),5.21(d,j=2.93hz,4h)7.73-8.03(m,11h)。
[0097]
化合物28的合成在氮气环境下向化合物27(17.4mg,7.79μmol)的dmf(2ml)溶液中添加三乙胺(8.7μl,62μmol)及三氟乙酸五氟苯酯(5.4μl,31μmol),将反应混合物在室温下搅拌1.5小时。在反应液中添加水,利用etoac进行萃取。利用饱和碳酸氢钠、硫酸氢钠、饱和食盐水洗净有机层。在减压下浓缩后,利用戊烷湿磨残渣,滤取并在40度、减压下加以干燥而获得化合物28
(11.72mg)。ms(esi)m/z:1200[m 2h]
1
h-nmr(600mhz,dmso-d6)δ(ppm):1.24(br s,20h),1.31-1.40(m,8h),1.41-1.49(m,8h),1.61-1.72(m,2h),1.77(s,12h),1.79-1.86(m,2h),1.89(s,12h),1.99(s,12h),2.01-2.07(m,2h),2.10(s,12h),2.12-2.21(m,4h),2.25-2.31(m,2h),2.80(br t,j=7.34hz,2h),2.95-3.08(m,8h),3.37-3.43(m,4h),3.65-3.72(m,4h),3.82-3.90(m,4h),3.97-4.06(m,12h),4.10-4.22(m,3h),4.48(d,j=8.44hz,4h),4.97(dd,j=11.00,2.93hz,4h),5.21(d,j=2.93hz,4h),7.70-7.86(m,8h),7.86-8.01(m,3h)。
[0098]
实施例1~7:核酸复合体1~7的合成seq-1及seq-2是委托jeen design公司合成。在seq-2(1.3μmol)中添加四硼酸钠缓冲液(sodium tetraborate buffer)(ph值为8.5,最终浓度40mm),并添加溶解于dmso中的dbco-nhs酯(dibenzocyclooctyne-n-hydroxysuccinimidyl ester,二苯并环辛炔-n-羟基丁二酰亚胺酯)(cas no.1353016-71-3,60μmol),在室温下搅拌15分钟。在反应液中添加水后,利用pd-10管柱(ge healthcare公司)进行凝胶过滤纯化。进而利用amicon ultra 3k(millipore公司)进行纯化、浓缩,由此获得粗产物(seq-3)。在粗产物(40nmol)中添加乙酸三乙基铵(triethylammonium acetate)(ph值为7.0,最终浓度50mm),并分别添加溶解于水中的化合物8、11、14、15、17、23、24(400nmol),在室温下搅拌15分钟。利用pd-10管柱(ge healthcare公司)对反应液进行凝胶过滤纯化。进而利用amicon ultra3k(millipore公司)进行纯化、浓缩,由此获得核酸复合体、即对应于化合物8、11、14、15、17、23、24的核酸复合体1~7。
[0099][0100]
核酸复合体1
[0101]
核酸复合体2
[0102]
核酸复合体3
[0103]
核酸复合体4
[0104]
核酸复合体5
[0105]
核酸复合体6
[0106]
核酸复合体7
[0107]
实施例8:核酸复合体8的合成在seq-2(26.6nmol)中添加四硼酸钠缓冲液(ph值为8.5)及溶解于dmso中的化合物28(120nmol),在室温下进行搅拌。在反应液中添加水后,利用amicon ultra 3k(millipore公司)进行纯化。在所获得的粗产物中添加5倍量的28%氨水,在室温下静置3小时。在反应液中添加水后,利用amicon ultra 3k(millipore公司)进行纯化。进而通过反相hplc进行纯化,由此获得核酸复合体8。
[0108]
核酸复合体8
[0109]
核酸序列的说明本实施例所使用的核酸的序列中(5'-3')为a(f)^g(m)^g(f)a(m)c(f)u(m)g(f)g(m)u(f)c(m)u(f)u(f)u(m)c(f)u(m)a(f)u(m)a(f)u(m)^c(f)^u(m),大写字母表示rna,(m)表示2'-o-甲基修饰rna,(f)表示2'-氟rna,^表示硫代磷酸酯键。又,在各寡核苷酸的5'端结合有作为荧光色素的花青色素cy3(激发波长555nm,荧光波长570nm)。对seq-1、2及本实施例中所合成的核酸复合体进行利用maldi-tof-ms的分子量测定,将其结果示于表1。
[0110]
[表1]
[0111]
<试验例1>核酸复合体对人cd206表达lenti-x 293t细胞的活体外吸收活性评估对于实施例中所合成的核酸复合体,分别通过以下方法导入人cd206表达lenti-x 293t细胞(clontech公司),对吸收进行评估。将人cd206表达lenti-x 293t细胞(clontech公司)以成为2
×
104个/100μl/孔的方式接种至96孔pdl(poly-d-lysine,聚赖氨酸)涂层板(corning公司),在37℃的5%co2培养箱内培养2天。以最终浓度成为100nmol/l的方式,添加seq-1或所合成的核酸复合体,在37℃的5%co2培养箱内培养2小时。添加包含10μg/ml hoechst(life technologies公司)的4%多聚甲醛/pbs(phosphate buffered saline,磷酸盐缓冲盐水)(wako),在常温下将细胞固定30分钟,利用pbs洗净4次。利用in cell analyzer 2200(ge healthcare公司)进行荧光成像解析,利用核数修正孔内的cy3荧光强度,算出每个细胞的平均cy3荧光强度。此时,将添加了未经糖配体修饰的核酸seq-1的孔的荧光强度设为1,以相对值的形式算出添加了各核酸复合体的孔的荧光强度。
[0112]
将结果示于表2。根据该结果可知,与不含糖配体的seq-1相比,含有galnac的核酸复合体6未被有效率地吸收至cd206表达的细胞中,另一方面,含有甘露糖的核酸复合体1至5被有效率地吸收至cd206表达的细胞中。
[0113]
[表2]
[0114]
<试验例2>核酸复合体对人asgr1表达lenti-x293t细胞的活体外吸收活性评估对于实施例中所合成的核酸复合体,分别通过以下方法导入至人asgr1表达lenti-x293t细胞(clontech公司)中,对吸收活性进行评估。将人asgr1表达lenti-x293t细胞(clontech公司)以成为2
×
104个/100μl/孔的方式接种至96孔pdl涂层板(corning公司),在37℃的5%co2培养箱内培养1天或2天。以最终浓度成为100nmol/l的方式,添加seq-1或所合成的核酸复合体,在37℃的5%co2培养箱内培养2小时。其后的荧光成像解析是通过与试验例1同样的方法实施。
[0115]
将结果示于表3。根据该结果可知,与不含糖配体的seq-1相比,含有甘露糖的核酸复合体1至5未被有效率地吸收至asgr1表达的细胞中,另一方面,含有galnac的核酸复合体6至8被有效率地吸收至asgr1表达的细胞中。
[0116]
[表3]
[0117]
<试验例3>小鼠肝实质细胞及库普弗细胞(kupffer cell)中的核酸复合体的吸收评估(活体内评估)分别以1mg/kg对balb/c小鼠(n=3)皮下施用seq-1、核酸复合体3及6。施用4小时后,对肝脏进行灌流,利用流式细胞仪将肝实质细胞及库普弗细胞分离,对荧光强度进行测定。此时,将施用了seq-1的组的荧光强度设为1,以相对值的形式算出各核酸复合体施用组的荧光强度。
将结果示于表4。根据该结果可知,与不含糖配体的seq-1相比,含有甘露糖的核酸复合体3被高效地吸收至报告为cd206阳性的小鼠库普弗细胞中,含有galnac的核酸复合体6被高效地吸收至报告为asgr1阳性的小鼠肝实质细胞中。
[0118]
[表4]
[0119]
化合物37的合成流程
[0120]
化合物30的合成
[0121]
在50℃加热乙酸肼(1.29g,14.06mmol)与dmf(50ml)的混合物。继而,将其在15℃的水浴中冷却,添加α-d-甘露糖五乙酸酯(4.99g,12.8mmol)。将所获得的溶液在15℃-19℃的水浴中搅拌16小时。在混合物中添加水与etoac进行萃取,利用etoac进一步萃取水层。利用饱和食盐水洗净合并的有机层,利用无水硫酸钠加以干燥并过滤,在减压下进行浓缩。通过使用biotage isolera(100g kp-sil,20-100%etoac/环己烷)的管柱色谱法纯化残渣而以α及β非镜像异构物的9:1混合物的形式获得化合物30(4.19g)。α异构物1h nmr(600mhz,cdcl3)δppm:2.00(s,3h),2.05(s,3h),2.11(s,3h),2.16(s,3h),3.00(d,j=4.03hz,1h),4.15(br d,j=10.64hz,1h),4.22-4.28(m,2h),5.23-5.33(m,3h),5.43(br dd,j=10.27,3.30hz,1h)
[0122]
化合物31的合成在氮气环境下向化合物30(3.64g,10.5mmol)的dcm(218ml)溶液中滴加三氯乙腈(10.48ml,104.5mmol),继而滴加dbu(0.158ml,1.05mmol)。将混合物在氮气环境、室温下搅拌2.25小时。在减压下进行浓缩,通过使用biotage isolera(100g kp-sil,0-50%etoac/环己烷)的管柱色谱法纯化残渣而获得化合物31(4.567g)。1h nmr(600mhz,cdcl3)δppm:2.01(s,3h),2.07(s,3h),2.08(s,3h),2.20(s,3h),4.14-4.23(m,2h),4.28(dd,j=12.47,4.77hz,1h),5.38-5.43(m,2h),5.47(br s,1h),6.28(s,1h),8.79(s,1h)
[0123]
化合物33的合成
[0124]
将化合物31(2.45g,4.96mmol)及n-cbz-6-胺基-己烷-1-醇(1.87g,7.45mmol)的dcm(90ml)溶液冷却为2℃(内部温度),在氮气环境下滴加三氟化硼合二乙醚(0.126ml,
0.993mmol)。将所获得的混合物在2℃搅拌25分钟,继而在室温下搅拌1小时20分钟。在混合物中添加饱和碳酸氢钠水溶液进行萃取,利用dcm萃取水层。利用饱和食盐水洗净合并的有机层,利用疏水性滤纸加以过滤,并在减压下进行浓缩。通过使用biotage isolera(100g kp-sil,0-100%etoac/环己烷)的管柱色谱法纯化残渣而获得化合物33(1.37g)。1h nmr(600mhz,cdcl3)δppm:1.32-1.43(m,4h),1.49-1.53(m,2h),1.56-1.64(m,2h),1.99(s,3h),2.04(s,3h),2.10(s,3h),2.15(s,3h),3.20(q,j=6.72hz,2h),3.40-3.48(m,1h),3.64-3.71(m,1h),3.97(ddd,j=9.90,5.32,2.38hz,1h),4.08-4.12(m,1h),4.28(dd,j=12.29,5.32hz,1h),4.79(d,j=1.47hz,2h),5.10(s,2h),5.22(dd,j=3.30,1.83hz,1h),5.28(t,j=9.54hz,1h),5.34(dd,j=10.64,3.67hz,1h),7.29-7.33(m,1h),7.36(d,j=4.03hz,4h)
[0125]
化合物34的合成
[0126]
在氮气环境下向化合物33(1.35g,2.31mmol)的ims(38ml)溶液中添加10%钯-碳(0.293g,50%含水品),在氢气环境下在室温下搅拌1.5小时。利用硅藻土(注册商标)过滤混合物,并利用ims洗净。在滤液中添加4n氯化氢的1,4-二噁烷溶液(0.78ml,3.12mmol),在减压下进行浓缩而获得化合物34(1.06g)。1h nmr(600mhz,dmso-d6)δppm:1.30-1.37(m,4h),1.51-1.61(m,4h),1.94(s,3h),2.03(s,6h),2.11(s,3h),2.72-2.80(m,2h),3.42-3.50(m,1h),3.63(dt,j=9.72,6.88hz,1h),3.88-3.96(m,1h),4.06(dd,j=12.10,2.57hz,1h),4.15(dd,j=12.29,5.32hz,1h),4.87(s,1h),5.04-5.15(m,3h),7.80(br s,3h)
[0127]
化合物35的合成
[0128]
在化合物34(510mg,1.05mmol)及化合物25(128.4mg,0.21mmol)的dmf(3ml)溶液中依序添加hobt(161mg,1.05mmol)及edci(202mg,1.05mmol)。将所获得的混合物在室温下搅拌10分钟,继而添加dipea(0.18ml,1.05mmol)。将所获得的溶液在室温下搅拌22小时。追加化合物34(505.9mg,1.04mmol)的dmf(1.5ml)溶液、hobt(161mg,1.05mmol)及edci
(202mg,1.05mmol),10分钟后添加dipea(0.18ml,1.05mmol)。将混合物进一步搅拌3小时。通过制备型hplc纯化混合物而获得化合物34(46.3mg)。1h nmr(600mhz,dmso-d6)δppm:1.21-1.45(m,24h),1.49-1.59(m,8h),1.61-1.89(m,8h),1.93(s,12h),2.01(s,24h),2.10(s,12h),2.07-2.23(m,8h),2.33-2.37(m,2h),2.96-3.08(m,7h),3.41-3.48(m,4h),3.57-3.65(m,4h),3.88-3.94(m,4h),4.05(dd,j=12.10,1.83hz,4h),4.10-4.20(m,8h),4.85(s,4h),5.06-5.13(m,14h),7.31-7.40(m,6h),7.69-7.94(m,6h);lcms(esi ):m/z 1164.58[m 2h]
2
。
[0129]
化合物36的合成
[0130]
在氮气环境下向化合物35(40.7mg,0.02mmol)的ims(6ml)溶液中添加10%钯-碳(11.5mg,50%含水品),在氢气环境下在室温下搅拌2小时。利用硅藻土(注册商标)过滤混合物,并利用ims洗净。将合并的滤液在减压下进行浓缩而获得化合物36(43.8mg)。1h nmr(600mhz,dmso-d6)δppm:1.20-1.44(m,24h),1.51-1.60(m,8h),1.61-1.88(m,8h),1.93(s,12h),2.02(s,12h),2.02(s,12h),2.10(s,12h),2.12-2.27(m,10h),2.96-3.08(m,7h),3.41-3.48(m,4h),3.58-3.65(m,4h),3.88-3.94(m,4h),4.04(br d,j=10.27hz,4h),4.09-4.29(m,8h),4.85(s,4h),5.04-5.14(m,12h),7.71-8.01(br m,7h)
[0131]
化合物37的合成
[0132]
在化合物36(32.8mg,0.015mmol)的dmf(3ml)溶液中添加三甲胺(16.3μl,0.12mmol),继而滴加三氟乙酸五氟苯酯(10.0μl,0.06mmol),将所获得的混合物在室温下搅拌1.25小时。将反应混合物注入至水中,并添加饱和食盐水,并利用etoac进行萃取。利用饱和碳酸氢钠水溶液、1m硫酸氢钠水溶液及饱和食盐水依序洗净有机层。利用无水硫酸钠
使有机层干燥并进行过滤,在减压下进行浓缩。利用戊烷湿磨残渣。继而,将固体在40℃减压干燥而获得化合物37(36.7mg)。1h nmr(600mhz,dmso-d6)δppm:1.20-1.47(m,24h),1.51-1.61(m,8h),1.62-1.91(m,8h),1.93(s,12h),2.01(s,24h),2.10(s,12h),2.12-2.33(m,8h),2.80(br t,j=6.79hz,2h),2.96-3.09(m,7h),3.41-3.50(m,4h),3.58-3.65(m,4h),3.91(br s,4h),4.04(br d,j=12.10hz,4h),4.10-4.28(m,8h),4.85(br s,4h),5.06-5.15(m,12h),7.68-8.03(m,7h);lcms(esi ):m/z 1202.46[m 2h]
2
。
[0133]
化合物44的合成流程
[0134]
化合物39的合成
[0135]
在氩气下称量无水氯化锌(3.65g,26.8mmol)到250ml的三口烧瓶中。将烧瓶在减
压下在110℃干燥1小时,继而在减压下放置冷却一晚。继而,使烧瓶充满氩气,添加三乙酸(2s,3r,4r,5r,6r)-3-乙酰胺-6-(乙酰氧基甲基)四氢-2h-吡喃-2,4,5-三基酯(7.85g,20.2mmol)、n-cbz-6-胺基-己烷-1-醇(6.74g,26.8mmol)及dce(85ml)。将混合物在氩气下在70℃(外部温度)加热3小时后,将混合物冷却,继而利用乙酸乙酯及饱和碳酸氢钠水溶液进行稀释。将混合物搅拌20分钟,利用硅藻土(注册商标)进行过滤,并利用乙酸乙酯洗净。分配合并的滤液,利用水、继而利用饱和食盐水洗净有机层。利用无水硫酸钠使有机层干燥并进行过滤,在减压下进行浓缩。通过使用combiflash的管柱色谱法(330g puriflash si,20-100%etoac/环己烷)纯化残渣而获得化合物39(9.42g)。1h nmr(400mhz,cdcl3)δppm:1.30-1.43(m,4h),1.45-1.65(m,4h),1.94(s,3h),2.00(s,3h),2.05(s,3h),2.13(s,3h),3.12-3.29(m,2h),3.48(dt,j=9.63,6.50hz,1h),3.79-4.02(m,3h),4.06-4.21(m,2h),4.65(br d,j=8.19hz,1h),4.78-4.93(m,1h),5.06-5.18(m,2h),5.27(dd,j=11.25,3.06hz,1h),5.34(d,j=2.81hz,1h),5.77(br d,j=8.44hz,1h),7.29-7.42(m,5h)lcms(m/z 581[m h]
)
[0136]
化合物40的合成
[0137]
在氮气环境下向化合物39(9.23g,15.9mmol)的ims(275ml)溶液中添加4m氯化氢的二噁烷溶液(5.17ml,20.7mmol)及10%钯-碳(1.895g,50%含水品),在氢气环境下在室温下搅拌2.5小时。利用硅藻土(注册商标)过滤混合物,并利用ims洗净。在减压下浓缩滤液而获得化合物40(8.07g)。1h nmr(400mhz,dmso-d6)δppm:1.22-1.35(m,4h),1.39-1.62(m,4h),1.78(s,3h),1.89(s,3h),2.00(s,3h),2.10(s,3h),2.69-2.82(m,2h),3.41-3.47(m,2h),3.64-3.77(m,1h),3.81-3.93(m,1h),3.95-4.12(m,2h),4.49(d,j=8.56hz,1h),4.97(dd,j=11.25,3.42hz,1h),5.22(d,j=3.42hz,1h),7.62-7.80(m,3h),7.86(d,j=9.29hz,1h)lcms(m/z 447[m h]
)
[0138]
化合物41的合成
[0139]
在250ml的quickfit(注册商标)三角烧瓶中的ctc树脂(1.33mmol/g,1.31g,1.74mmol)、fmoc-glu(otbu)-oh(0.370g,0.87mmol)、fmoc-d-glu(otbu)-oh(0.370g,0.87mmol)及dcm(105ml)的混合物中滴加dipea(1.215ml,6.96mmol)。塞住烧瓶,安装于盘式振荡器,以300rpm振荡2小时。添加meoh(0.7ml,17.3mmol),将混合物进一步振荡35分钟。
利用70ml塑料相分离筒过滤混合物,通入氮气并利用20%哌啶的dmf溶液(50ml)处理30分钟。排出溶液,利用dmf洗净树脂。在筒中的树脂中添加fmoc-d-glu-otbu(0.740g,1.74mmol)、n-fmoc-l-谷氨酸-1-叔丁酯(0.740g,1.74mmol)及hbtu(1.253g,3.30mmol)的dmf(10ml)溶液,继而滴加dipea(1.22ml,6.96mmol)。对混合物通入氮气并混合1小时。继而,利用氮气气流将溶液的溶剂去除,通过通入氮气混合树脂,并利用20%哌啶dmf(50ml)溶液处理35分钟。排出溶液,利用dmf洗净树脂。在筒中的剩余的树脂中滴加1,5-戊二酸单苄基酯(0.580g,2.61mmol)及hbtu(1.253g,3.30mmol)的dmf(10ml)溶液,继而滴加dipea(1.215ml,6.96mmol)。对混合物通入氮气80分钟进行混合。排出溶液,利用dmf、继而利用meoh洗净树脂。继而,将树脂进行抽吸干燥。将树脂转移至25
×
150mm试管中,减压干燥30分钟后,利用dcm(5ml)及tfa(5ml)进行处理,并利用盘式振荡器振荡2小时。继而,将混合物通过相分离筒进行过滤,并利用dcm洗净。在减压下浓缩滤液而获得化合物41(0.5859g)。1h nmr(400mhz,dmso-d6)δppm:1.68-1.84(m,4h),1.86-2.01(m,2h),2.13-2.23(m,4h),2.23-2.29(m,2h),2.38(t,j=7.52hz,2h),4.11-4.22(m,2h),5.09(s,2h),7.29-7.42(m,5h),8.06-8.17(m,2h)lcms(m/z 481[m h]
)
[0140]
化合物42的合成
[0141]
在化合物40(4.06g,8.41mmol)及化合物41(0.5821g,1.21mmol)的dmf(8ml)溶液中依序添加hobt(1.392g,9.09mmol)、edci(1.742g,9.09mmol),继而添加dipea(1.587ml,9.09mmol)。将混合物在室温下搅拌20小时。通过制备型hplc纯化混合物而获得化合物42(0.48g)。1h nmr(600mhz,dmso-d6)δppm:1.23(br s,12h),1.29-1.40(m,6h),1.40-1.48(m,6h),1.60-1.71(m,2h),1.76(s,9h),1.76-1.87(m,4h),1.89(s,9h),1.99(s,9h),2.00-2.07(m,2h),2.10(s,9h),2.11-2.15(m,2h),2.18(br t,j=7.15hz,2h),2.34-2.38(m,2h),2.94-3.07(m,6h),3.36-3.43(m,3h),3.65-3.73(m,3h),3.82-3.91(m,3h),3.98-4.06(m,9h),4.10-4.20(m,2h),4.48(d,j=8.44hz,3h),4.97(dd,j=11.19,3.12hz,3h),5.08(s,2h),5.21(d,j=3.30hz,3h),7.30-7.39(m,5h),7.71-7.76(m,1h),7.77-7.85(m,5h),7.87-7.93(m,2h)lcms(m/z 1766[m h]
)
[0142]
化合物43的合成
4.06(m,9h),4.09-4.22(m,2h),4.48(d,j=8.44hz,3h),4.97(dd,j=11.37,3.30hz,3h),5.21(d,j=3.30hz,3h),7.73(br t,j=5.14hz,1h),7.77-7.85(m,5h),7.91(br dd,j=11.19,7.89hz,1h),7.98(br d,j=8.07hz,1h)lcms(m/z 1843.56[m h]
)
[0146]
化合物47的合成流程
[0147]
化合物45的合成
[0148]
在化合物34(1383mg,2.86mmol)及化合物41(183.1mg,0.381mmol)的dmf(3.0ml)溶液中依序添加hobt(438mg,2.86mmol)、edci(548mg,2.86mmol)及dipea(0.50ml,2.86mmol)。将所获得的混合物在室温下搅拌24小时。通过制备型hplc纯化混合物而获得化合物45(133.4mg)。1h nmr(600mhz,dmso-d6)δppm:1.23-1.34(m,12h),1.34-1.43(m,6h),1.50-1.59(m,6h),1.62-1.72(m,2h),1.74-1.80(m,2h),1.81-1.89(m,2h),1.93(s,9h),2.01(s,18h),2.03-2.07(m,2h),2.10(s,9h),2.11-2.15(m,2h),2.16-2.21(m,2h),2.33-2.38(m,2h),2.97-3.07(m,6h),3.41-3.48(m,3h),3.57-3.64(m,3h),3.87-3.94(m,3h),4.05(dd,j=12.10,2.57hz,3h),4.10-4.19(m,5h),4.85(s,3h),5.0-5.13(m,11h),7.29-7.39(m,5h),7.71(br t,j=4.95hz,1h),7.76-7.84(m,2h),7.85-7.91(m,2h)lcms(m/z 1769[m h]
)
[0149]
化合物46的合成
[0150]
在氮气环境下向化合物45(101.9mg,0.058mmol)的ims(20ml)溶液添加10%钯-碳(37.4mg,50%含水品)。在氢气环境下将混合物在室温下搅拌2.5小时。利用硅藻土(注册商标)过滤混合物,继而利用ims洗净。将合并的滤液在减压下浓缩而获得化合物46(95.9mg)。1h nmr(600mhz,dmso-d6)δppm:1.28(br s,12h),1.34-1.44(m,6h),1.49-1.60(m,6h),1.61-1.76(m,4h),1.81-1.90(m,2h),1.93(s,9h),2.02(s,9h),2.02(s,9h),2.04-2.08(m,2h),2.10(s,9h),2.12-2.23(m,6h),2.96-3.07(m,6h),3.40-3.48(m,3h),3.57-3.65(m,3h),3.86-3.94(m,3h),4.05(dd,j=12.10,2.38hz,3h),4.09-4.18(m,5h),4.85(s,3h),5.01-5.14(m,9h),7.62-8.17(m,5h),11.84-12.15(m,1h)。lcms(m/z 1679[m h]
)。
[0151]
化合物47的合成
[0152]
在氮气环境下向化合物46(94.6mg,0.056mmol)的dmf(9ml)溶液中添加三乙胺(62.8μl,0.451mmol),并滴加三氟乙酸五氟苯酯(38.7μl,0.225mmol)。将所获得的混合物在室温下搅拌1小时。将混合物注入至水中,并添加饱和食盐水,并利用etoac进行萃取。利用饱和碳酸氢钠水溶液、1m硫酸氢钠水溶液及饱和食盐水依序洗净合并的有机层。利用无水硫酸钠使有机层干燥并进行过滤,继而在减压下进行浓缩,由此获得化合物47(109.0mg)。1h nmr(600mhz,dmso-d6)δppm:1.23-1.33(m,12h),1.34-1.43(m,6h),1.50-1.60(m,6h),1.62-1.77(m,2h),1.80-1.91(m,4h),1.93(s,9h),2.01(s,18h),2.03-2.07(m,2h),2.10(s,9h),2.12-2.20(m,2h),2.25-2.30(m,2h),2.77-2.82(m,2h),2.96-3.08(m,6h),3.41-3.48(m,3h),3.53-3.68(m,3h),3.86-3.95(m,3h),4.00-4.07(m,3h),4.10-4.23(m,5h),4.85(s,3h)5.04-5.13(m,9h),7.71(br t,j=5.32hz,1h),7.78-7.85(m,2h),7.89(dd,j=12.84,8.07hz,1h),7.96(d,j=8.07hz,1h)
lcms(m/z 1845.47[m h]
)
[0153]
化合物53a、53b及53c的合成流程
[0154]
化合物49a的合成
[0155]
将装入50ml的三口烧瓶中的氯化锌(285mg,2.09mmol)在减压下在110℃干燥1小时,在减压下放置冷却一晚。对烧瓶进行氮气置换,添加化合物38(611mg,1.57mmol)及化合物48a(500mg,2.09mmol)的dce(5ml)溶液。在氮气环境下在70℃(外部温度)将混合物加热3小时。将混合物放置冷却,利用乙酸乙酯(10ml)/饱和碳酸氢钠水溶液(5ml)加以稀释。将混合物搅拌10分钟,利用硅藻土(注册商标)过滤,并利用乙酸乙酯洗净。利用水及饱和食盐水洗净合并的滤液。利用疏水性滤纸过滤有机层,并在减压下进行浓缩。通过使用biotage isolera(50g kp-sil,0-50%(3:1etoac-ims)/mtbe)的管柱色谱法纯化残渣,由此获得化合物49a(599.4mg)。1h nmr(600mhz,cdcl3)δppm:1.90(s,3h),2.00(s,3h),2.04(s,3h),2.13(s,3h),3.26-3.86(m,8h),3.87-4.00(m,2h),4.05-4.19(m,2h),4.76(br d,j=7.70hz,1h),5.10-5.20(m,2h),5.26(br dd,j=6.60,3.30hz,1h),5.32(br s,1h),5.43(br s,1h),5.75(br d,j=7.34hz,1h),7.28-7.42(m,5h)。
lcms(m/z 569[m h]
)。
[0156]
化合物50a的合成
[0157]
在氮气环境下向化合物49a(594.2mg,1.05mmol)的ims(18ml)溶液中添加4m氯化氢的1,4-二噁烷溶液(0.34ml,1.36mmol)及10%钯-碳(50%含水品,125mg)。在氢气环境下将混合物在室温下搅拌2.5小时。利用硅藻土(注册商标)过滤混合物,并利用ims洗净。将合并的滤液在减压下浓缩,由此获得化合物50a(507.9mg)。1h nmr(600mhz,dmso-d6)δppm:1.77-1.84(m,3h),1.86-1.92(m,3h),1.97-2.03(m,3h),2.06-2.13(m,3h),2.92-3.00(m,2h),3.52-3.75(m,5h),3.83(ddd,j=11.28,5.41,3.85hz,1h),3.86-3.93(m,1h),3.95-4.17(m,3h),4.57(d,j=8.44hz 1h),4.99(dd,j=11.19,3.48hz,1h),5.23(d,j=3.67hz,1h),7.81(br s,3h),7.85-7.92(m,1h)lcms(m/z 435[m h]
)
[0158]
化合物51a的合成
[0159]
在化合物50a(506.5mg,1.08mmol)及化合物41(68.9mg,0.14mmol)的dmf(2.0ml)溶液中依序添加hobt(165mg,1.08mmol)、edci(206mg,1.08mmol)及dipea(188μl,1.08mmol)。将所获得的混合物在室温下搅拌20小时。通过制备型hplc纯化混合物,由此获得化合物51a(40.0mg)。1h nmr(600mhz,dmso-d6)δppm 1.62-1.73(m,2h),1.77(s,9h),1.80-1.87(m,2h),1.89(s,9h),1.99(s,9h),2.02-2.08(m,2h),2.10(s,9h),2.11-2.24(m,4h),2.34-2.38(m,2h),3.13-3.22(m,4h),3.34-3.42(m,7h),3.44-3.54(m,7h),3.55-3.61(m,3h),3.73-3.80(m,3h),3.84-3.91(m,3h),3.97-4.08(m,9h),4.13-4.25(m,2h),4.55(dd,j=8.07,3.67hz,3h),4.99(br d,j=11.00hz,3h),5.07-5.10(m,2h),5.22(d,j=3.30hz,3h),7.31-7.40(m,5h),7.60-7.97(m,8h)lcms(m/z 1730/1731[m h]
)
[0160]
化合物52a的合成
[0161]
在氮气环境下向化合物51a(42.7mg,0.03mmol)的ims(8ml)溶液中添加10%钯-碳(50%含水品,16.0mg)。在氢气环境下将混合物在室温下搅拌2.5小时。利用硅藻土(注册商标)过滤混合物,并利用ims洗净。在减压下浓缩滤液而获得化合物52a(41.2mg)。1h nmr(600mhz,dmso-d6)δppm 1.62-1.75(m,4h),1.78(s,9h),1.81-1.87(m,2h),1.89(s,9h),2.00(s,9h),2.05-2.32(m,17h),3.12-3.23(m,4h),3.36-3.43(m,7h),3.45-3.54(m,7h),3.55-3.61(m,3h),3.74-3.81(m,3h),3.83-3.92(m,3h),3.98-4.08(m,9h),4.11-4.23(m,2h),4.51-4.59(m,3h),4.99(br d,j=11.00hz,3h),5.22(d,j=3.30hz,3h),7.62-8.04(m,8h)lcms(m/z 1640/1641[m h]
)
[0162]
化合物53a的合成
[0163]
在氮气环境下向化合物52a(41.6mg,0.03mmol)的dmf(4ml)溶液中滴加三乙胺(28μl,0.20mmol),继而滴加三氟乙酸五氟苯酯(17μl,0.10mmol)。将所获得的混合物在室温下搅拌1.5小时,注入至水中,添加饱和食盐水,并利用etoac进行萃取。利用饱和碳酸氢钠水溶液、1m硫酸氢钠水溶液及饱和食盐水依序洗净合并的有机层。利用无水硫酸钠使有机层干燥并进行过滤,在减压下进行浓缩。将残渣转移至装有dcm/meoh的样品瓶中,蒸发溶剂后,在40℃减压干燥2小时,由此获得化合物52c(27.1mg)。1h nmr(600mhz,dmso-d6)δppm:1.57-1.75(m,4h),1.77(s,9h),1.86-1.95(m,11h),2.00(s,9h),2.05-2.33(m,15h),2.77-2.88(m,2h),3.11-3.24(m,4h),3.41-3.54(m,14h),3.55-3.62(m,3h),3.74-3.81(m,3h),3.84-3.91(m,3h),3.98-4.07(m,9h),4.11-4.23(m,2h),4.55(br dd,j=8.07,3.67hz,3h),4.99(br d,j=11.00hz,3h),5.22(d,j=3.30hz,3h),7.65-8.04(m,8h)lcms(m/z 1806.83[m h]
)
[0164]
化合物49b的合成
[0165]
将装入50ml的三口烧瓶中的氯化锌(0.466g,3.42mmol)在减压下在110℃干燥95分钟,在减压下放置冷却一晚。对烧瓶进行氮气置换,并添加化合物38(1.00g,2.57mmol)及化合物48b(0.97g,3.42mmol)的dce(10ml)溶液。在氮气环境下在70℃(外部温度)将混合物加热3小时。将混合物放置冷却,利用乙酸乙酯(20ml)/饱和碳酸氢钠水溶液(10ml)加以稀释。将混合物搅拌15分钟,利用硅藻土(注册商标)过滤,并利用乙酸乙酯洗净。利用水及饱和食盐水洗净滤液。利用疏水性滤纸过滤有机层,并在减压下进行浓缩。通过使用biotage isolera(100g sfar duo si,0-50%(3:1etoac-ims)/mtbe)的管柱色谱法纯化残渣,由此获得化合物49b(1.14g)。1h nmr(600mhz,cdcl3)δppm:1.92(s,3h),1.98(s,3h),2.03(s,3h),2.14(s,3h),3.32-3.50(m,2h),3.53-3.73(m,8h),3.77-3.94(m,3h),4.02-4.22(m,3h),4.74(br d,j=8.44hz,1h),5.03-5.08(m,1h),5.11(br s,2h),5.26(br s,1h),5.40-5.53(m,1h),6.11-6.21(m,1h),7.30-7.40(m,5h)。lcms(m/z 613[m h]
)。
[0166]
化合物50b的合成
[0167]
在氮气环境下向化合物49b(1.14g,1.86mmol)的ims(32ml)溶液中添加4m氯化氢的1,4-二噁烷(0.60ml,2.42mmol)溶液及10%钯-碳(50%含水品,0.221g)。在氢气环境下将混合物在室温下搅拌2.5小时。利用硅藻土(注册商标)过滤混合物,并利用ims洗净。将合并的滤液在减压下浓缩而获得化合物50b(0.97g)。1h nmr(600mhz,dmso-d6)δppm 1.79(s,3h),1.89(s,3h),2.00(s,3h,)2.11(s,3h),2.94-3.02(m,2h),3.50-3.62(m,9h),3.77-3.84(m,1h),3.86-3.94(m,1h),3.98-4.10(m,3h),4.54(d,j=8.44hz,1h),4.97(dd,j=11.19,3.48hz,1h),5.22(d,j=3.67hz,1h),7.70-7.93(m,4h)lcms(m/z 479[m h]
)
[0168]
化合物51b的合成
[0169]
在化合物50b(556mg,1.08mmol)及化合物41(74.1mg,0.15mmol)的dmf(3ml)溶液中依序添加hobt(165mg,1.08mmol)、edci(207mg,1.08mmol)、及dipea(189μl,1.08mmol)。在室温下将所获得的混合物搅拌19小时。通过制备型hplc纯化混合物,由此获得化合物51b(67.5mg)。1h nmr(600mhz,dmso-d6)δppm 1.61-1.74(m,2h),1.77(s,9h),1.80-1.85(m,2h),1.89(s,9h),2.00(s,9h),2.03-2.08(m,2h),2.10(s,9h),2.12-2.24(m,4h),2.35-2.38(m,2h),3.10-3.23(m,4h),3.36-3.42(m,7h),3.44-3.63(m,22h),3.74-3.81(m,3h),3.83-3.91(m,3h),3.99-4.08(m,9h),4.13-4.25(m,2h),4.55(d,j=8.44hz,3h),4.95-5.00(m,3h),5.09(s,2h),5.21(d,j=3.30hz,3h),7.31-7.41(m,5h),7.72-7.86(m,4h),7.86-7.99(m,4h)lcms(m/z 1862/1863[m h]
)
[0170]
化合物52b的合成
[0171]
在氮气环境下向化合物51b(66.1mg,0.04mmol)的ims(13ml)溶液中添加10%钯-碳(50%含水品,23.1mg)。在氢气环境下将混合物在室温下搅拌2.5小时。利用硅藻土(注册商标)过滤混合物,并利用ims洗净。将合并的滤液在减压下浓缩而获得化合物52b(61.3mg)。1h nmr(600mhz,dmso-d6)δppm 1.63-1.75(m,4h),1.77(s,9h),1.81-1.87(m,2h),1.89(s,9h),2.00(s,9h),2.03-2.24(m,17h),3.09-3.23(m,4h),3.35-3.43(m,7h),3.44-3.64(m,22h),3.74-3.81(m,3h),3.84-3.91(m,3h),3.98-4.07(m,9h),4.10-4.24(m,2h),4.56(br d,j=8.80hz,3h),4.98(br d,j=11.00hz,3h),5.22(d,j=2.93hz,3h),7.70-8.06(m,8h)。lcms(m/z 1772/1773[m h]
)
[0172]
化合物53b的合成
[0173]
在氮气环境下向化合物52b(60.7mg,0.03mmol)的dmf(6ml)溶液中滴加三乙胺(38μl,0.27mmol),继而滴加三氟乙酸五氟苯酯(24μl,0.14mmol)。将所获得的混合物在室温下搅拌77分钟,注入至水中,添加饱和食盐水,并利用etoac进行萃取。利用饱和碳酸氢钠水溶液、1m硫酸氢钠水溶液、4%氯化锂水溶液及饱和食盐水依序洗净合并的有机层。利用无水硫酸钠使有机层干燥并进行过滤,继而进行浓缩,在40℃减压干燥4小时,由此获得化合物53b(29.3mg)。1h nmr(600mhz,dmso-d6)δppm:1.62-1.75(m,2h),1.77(s,9h),1.85-1.95(m,13h),2.00(s,9h),2.10(s,9h),2.12-2.33(m,6h),2.78-2.86(m,2h),3.12-3.24(m,4h),3.35-3.44(m,7h),3.45-3.64(m,22h),3.75-3.81(m,3h),3.83-3.92(m,3h),3.99-4.06(m,9h),4.12-4.26(m,2h),4.55(d,j=8.44hz,3h),4.98(dd,j=11.19,2.02hz,3h),5.21(d,j=3.67hz,3h),7.64-8.03(m,8h)lcms(m/z 1939.01[m h]
)
[0174]
化合物49c的合成
[0175]
将装入50ml的三口烧瓶中的氯化锌(210mg,1.54mmol)在减压下在110℃干燥75分钟,在减压下放置冷却一晚。对烧瓶进行氮气置换,添加化合物38(450.2mg,1.16mmol)及化合物48c(503.4mg,1.54mmol)的dce(5ml)溶液。在氩气环境下将混合物在70℃(外部温度)加热3小时。将混合物放置冷却,利用乙酸乙酯(10ml)/饱和碳酸氢钠水溶液(5ml)加以稀释。将混合物搅拌15分钟,利用硅藻土(注册商标)过滤,并利用乙酸乙酯洗净。利用水及饱和食盐水洗净滤液。利用疏水性滤纸过滤有机层,并在减压下进行浓缩。通过使用biotage isolera(50g sfar si,0-50%(3:1etoac-ims)/mtbe)的管柱色谱法纯化残渣,由此获得化合物49c(469.0mg)。1h nmr(600mhz,cdcl3)δppm:1.95(s,3h),1.96(s,3h),2.04(s,3h),2.15(s,3h),3.39(br d,j=4.77hz,2h),3.52-3.72(m,12h),3.79-3.94(m,3h),4.09-4.14(m,1h),4.14-4.18(m,1h),4.18-4.25(m,1h),4.77(br d,j=8.44hz,1h),4.98-5.06(m,1h),5.10(br s,2h),5.31(br s,1h),5.34-5.45(m,1h),6.29-6.39(m,1h),7.29-7.33(m,1h),7.36
(d,j=4.40hz,4h)lcms(m/z 657[m h]
)
[0176]
化合物50c的合成
[0177]
在氮气环境下向化合物49c(0.45g,0.69mmol)的ims(12ml)溶液中添加4m氯化氢的1,4-二噁烷溶液(225μl,0.90mmol)及10%钯-碳(50%含水品,0.083g)。在氢气环境下将混合物在室温下搅拌2.5小时。利用硅藻土(注册商标)过滤混合物,并利用ims洗净。在减压下浓缩滤液而获得化合物50c(0.40g)。1h nmr(600mhz,dmso-d6)δppm 1.78(s,3h),1.89(s,3h),2.00(s,3h),2.11(s,3h),2.94-3.01(m,2h),3.48-3.62(m,13h),3.76-3.82(m,1h),3.85-3.93(m,1h),3.99-4.08(m,3h),4.55(d,j=8.44hz,1h),4.94-5.01(m,1h),5.22(d,j=3.67hz,1h),7.75-7.88(m,4h)lcms(m/z 423[m h]
)
[0178]
化合物51c的合成
[0179]
在化合物50c(379.8mg,0.679mmol)及化合物41(47.2mg,0.10mmol)的dmf(2ml)溶液中依序添加hobt(104mg,0.68mmol)、edci(130mg,0.68mmol)及dipea(119μl,0.68mmol)。在室温下将所获得的混合物搅拌19小时。通过制备型hplc纯化混合物,由此获得化合物51c(53.8mg)。1h nmr(600mhz,dmso-d6)δppm 1.61-1.73(m,2h),1.77(s,9h),1.79-1.85(m,2h),1.89(s,9h),2.00(s,9h),2.03-2.08(m,2h),2.10(s,9h),2.12-2.22(m,4h),2.33-2.37(m,2h),3.07-3.23(m,4h),3.34-3.42(m,7h),3.43-3.55(m,30h),3.55-3.62(m,4h),3.75-3.81(m,3h),3.85-3.92(m,3h),3.99-4.07(m,9h),4.13-4.24(m,2h),4.56(d,j=8.80hz,3h),4.97(dd,j=11.00,3.30hz,3h),5.06-5.11(m,2h),5.21(d,j=3.30hz,3h),7.30-7.39(m,5h),7.62-7.84(m,5h),7.85-7.95(m,3h)lcms(m/z 1994/1995[m h]
)
[0180]
化合物52c的合成
[0181]
在氮气环境下向化合物51c(84mg,0.04mmol)的ims(17ml)溶液中添加10%钯-碳(50%含水品,27.3mg)。在氢气环境下将混合物在室温下搅拌2.5小时。利用硅藻土(注册商标)过滤混合物,并利用ims洗净。在减压下浓缩滤液而获得化合物52c(78.9mg)。1h nmr(600mhz,dmso-d6)δppm 1.60-1.75(m,4h),1.78(s,9h),1.80-1.87(m,2h),1.89(s,9h),2.00(s,9h),2.03-2.32(m,17h),3.12-3.23(m,4h),3.36-3.42(m,7h),3.43-3.63(m,34h),3.74-3.81(m,3h),3.84-3.92(m,3h),3.99-4.07(m,9h),4.09-4.22(m,2h),4.57(br d,j=8.44hz,3h),4.98(dd,j=11.19,3.12hz,3h),5.21(d,j=3.30hz,3h),7.61-8.05(m,8h),12.01(br s,1h)lcms(m/z 1904/1905[m h]
)
[0182]
化合物53c的合成
[0183]
在氮气环境下向化合物52c(77.3mg,0.04mmol)的dmf(3ml)溶液中滴加三乙胺(45μl,0.33mmol),继而滴加三氟乙酸五氟苯酯(27μl,0.16mmol)。将所获得的混合物在室温下搅拌1.5小时,注入至水中,添加饱和食盐水,继而利用etoac进行萃取。利用饱和碳酸氢钠水溶液、1m硫酸氢钠水溶液、继而利用饱和食盐水依序洗净有机层。利用无水硫酸钠使有机层干燥并进行过滤,继而在减压下进行浓缩。将残渣转移至装有dcm/meoh的样品瓶中,蒸发溶剂后,在40℃减压干燥2小时而获得残渣。将其溶解于etoac(20ml)中,利用4%氯化锂水溶液(5ml)、继而利用饱和食盐水(5ml)进行洗净,利用无水硫酸钠加以干燥并过滤,继而使其蒸发。将残渣转移至装有etoac的样品瓶中使其蒸发,在40℃减压干燥。将dcm添加至残渣中,在减压下进行浓缩,将残渣在40℃减压干燥4小时,由此获得化合物53c(19.4mg)。1h nmr(600mhz,dmso-d6)δppm:1.59-1.73(m,2h),1.77(s,9h),1.85-1.95(m,13h),2.00(s,9h),2.10(s,9h),2.12-2.34(m,6h),2.77-2.89(m,2h),3.11-3.23(m,4h),3.35-3.44(m,7h),3.45-3.63(m,34h),3.75-3.80(m,3h),3.84-3.91(m,3h),4.00-4.07(m,9h),4.16-4.26(m,2h),4.56(d,j=8.80hz,3h),4.97(dd,j=11.37,3.30hz,3h),5.21(d,j
=3.67hz,3h),7.62-8.03(m,8h)。qc lcms(m/z 1035.97[m 2h]
2
)
[0184]
实施例9~12:核酸复合体9~12的合成在b2m(β2-微球蛋白)靶sirna的正义链(215nmol)中添加四硼酸钠缓冲液(ph值为8.5)及溶解于dmso中的化合物28或44(1000nmol),在室温下进行搅拌。在反应液中添加水后,利用vivaspin3k(sartorius公司)进行纯化。在所获得的粗产物中添加5倍量的28%氨水,在室温下静置2小时。在反应液中添加水后,利用vivaspin3k(sartorius公司)反复纯化,由此获得核酸复合体9~12。通过esi-ms或maldi-tof-ms进行本实施例中所合成的核酸复合体的分子量。将所使用的正义链的序列、反义链的序列及分子量示于表5。
[0185]
[表5]
[0186]
将核酸复合体的序列及分子量示于表6。
[0187]
[表6]
[0188]
核酸复合体9
[0189]
核酸复合体10
[0190]
核酸复合体11
[0191]
核酸复合体12
[0192]
实施例13~16:核酸复合体13~16(si-rna)的制备等摩尔量添加反义链(sib2m-as)与核酸复合体9~12,由此制备以b2m为靶的si-rna。在表7中示出si-rna的正义链及反义链。
[0193]
[表7]
[0194]
<试验例4>将实施例13~16中所制备的核酸复合体(si-rna)以1.0mg/kg或5.0mg/kg皮下施用至balb/c小鼠(每组3只),施用起7天后取下肝脏。使用taqman探针(applied biosystems公司),通过qpcr(quantitative polymerase chain reaction,定量聚合酶链反应)测定肝脏中的b2m及作为内部对照的gapdh(甘油醛-3-磷酸脱氢酶)的mrna表达量。将无处理组(对照组)的小鼠肝脏中的b2m mrna表达量设为100%,算出核酸复合体施用组的b2m mrna表达量(相对值)。将结果示于表8。
[0195]
[表8]
[0196]
根据表8所示的结果可知,sirna-2、sirna-3、sirna-4及sirna-5表现出用量依赖性的基因表达抑制效果。不含糖配体的sirna-1在5.0mg/kg时也大致未见基因表达抑制效果,与此相对,含有糖配体的sirna在1.0mg/kg时也表现出较高的基因表达抑制效果。
[0197]
实施例17~19:核酸复合体17~19的合成使用具有3-10-3结构的gapmer型lna反义核酸(aso;isis-549148)作为核酸,并使用化合物44、47或53a,除此以外,通过与实施例9~11同样的方法制备核酸复合体。将所使用的正义链的序列、反义链的序列及分子量示于表9。
[0198]
[表9]5(l):lna 5-甲基胞嘧啶;5(x)=dna 5-甲基胞嘧啶
[0199]
将核酸复合体的序列及分子量示于表10。
[0200]
[表10]
[0201]
核酸复合体17
[0202]
核酸复合体18
[0203]
核酸复合体19
[0204]
<试验例5>核酸复合体对人cd206表达lenti-x 293t细胞的活体外吸收活性评估以与试验例1同样的方式进行核酸复合体17~19的评估。在试验例5中,将添加有未经糖配体修饰的核酸aso_3'cy3的孔的荧光强度设为1,以相对值的形式算出添加有各核酸复合体的孔的荧光强度。
[0205]
将结果示于表11。根据该结果可知,与不含糖配体的核酸aso_3'cy3相比,含有galnac的核酸复合体17及19未被有效率地吸收至cd206表达的细胞中,另一方面,含有甘露糖的核酸复合体18被有效率地吸收至cd206表达的细胞中。
[0206]
[表11]
[0207]
<试验例6>核酸复合体对人asgr1表达lenti-x293t细胞的活体外吸收活性评估以与试验例2同样的方式进行核酸复合体17~19的评估。在试验例6中,将添加有未经糖配体修饰的核酸aso_3'cy3的孔的荧光强度设为1,以相对值的形式算出添加有各核酸复合体的孔的荧光强度。
[0208]
将结果示于表12。根据该结果可知,与未经糖配体修饰的核酸aso_3'cy3相比,含有甘露糖的核酸复合体18未被有效率地吸收至asgr1表达的细胞中,另一方面,含有galnac的核酸复合体17及19被有效率地吸收至asgr1表达的细胞中。
[0209]
[表12]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
