制备3,6-二取代咪唑并[1,2-b]哒嗪衍生物的方法
[0001]
本技术是中国专利申请号201880017847.7(pct/jp2018/00959 6),申请日2018年03月13日,发明名称为“制备3,6-二取代咪唑并[1,2-b]哒嗪衍生物的方法”的分案申请。
技术领域
[0002]
本发明涉及制备3,6-二取代咪唑并[1,2-b]哒嗪衍生物的新方法。
背景技术:
[0003]
已知3,6-二取代咪唑并[1,2-b]哒嗪衍生物可用作药物或制备药物的起始材料,并且也可用于治疗肿瘤(专利文献1)。
[0004]
在专利文献1中,所有3,6-二取代咪唑并[1,2-b]哒嗪衍生物通过以下合成:使用芳香族亲核取代反应,将取代基引入作为起始材料的3-溴-6-氯咪唑并[1,2-b]哒嗪的6位,然后使用suzuki-miyaura偶联反应将取代基引入其3位(专利文献1,例如,实施例21)。
[0005]
此外,作为合成3,6-二取代咪唑并[1,2-b]哒嗪衍生物的另一种方法,已知一种方法,该方法包括使用利用钯催化的c-h活化的芳族取代反应,将芳基引入作为起始材料的6-氯咪唑并[1,2-b]哒嗪的3位。(非专利文献1)。
[0006]
专利文献1的合成方法具有以下限制:卤素原子对于咪唑并[1,2-b]哒嗪环上的suzuki-miyaura偶联反应的反应位点是必需的。另一方面,非专利文献1的合成方法的缺点在于需要大量的钯催化剂,并且当在要引入的芳基上存在给电子取代基时,产率变为中等程度(非专利文献1,例如,表2条目11)。
[0007]
引用列表
[0008]
专利文献
[0009]
专利文献1:wo2013-183578
[0010]
非专利文献
[0011]
非专利文献1:eur.j.org.chem.,862-871(2010)
技术实现要素:
[0012]
技术问题
[0013]
本发明提供了制备3,6-二取代咪唑并[1,2-b]哒嗪衍生物的方法,所述方法使用利用钯催化的c-h活化的芳族取代反应,使用6-氟咪唑并[1,2-b]哒嗪作为起始材料,其中该方法是一种行业上有用的新方法,其中使用的钯催化剂的量很小,并且具有复杂结构的电子供体取代基的芳基可以高产率被引入。
[0014]
问题的解决方案
[0015]
本发明涉及以下(1)至(7)。
[0016]
(1)制备式(iii)表示的化合物或其盐的方法:
[0017]
[式3]
[0018][0019]
其中pg表示氮原子的保护基团,
[0020]
该方法包括在钯催化剂和碱存在下在溶剂中,使式(i)表示的化合物或其盐:
[0021]
[式1]
[0022][0023]
与式(ii)表示的化合物或其盐反应:
[0024]
[式2]
[0025][0026]
其中每个符号如上所定义。
[0027]
(2)根据上述(1)的方法,其中在式(ii)和(iii)中,pg是叔丁氧基羰基。
[0028]
(3)根据上述(1)或(2)的方法,其中钯催化剂是由乙酸钯和三(2-甲基苯基)膦构成的催化剂。
[0029]
(4)根据上述(1)~(3)中任一项的方法,其中,碱为碳酸钾。
[0030]
(5)根据上述(1)~(4)中任一项的方法,其中,溶剂为二乙二醇二甲醚。
[0031]
(6)制备式(v)表示的化合物或其盐的方法:
[0032]
[式5]
[0033][0034]
该方法包括:
[0035]
使通过根据上述(1)~(5)中任一项的方法制备的式(iii)表示的化合物或其盐与式(iv)表示的化合物或其盐反应的步骤:
[0036]
[式4]
[0037][0038]
和
[0039]
使氮原子上的pg脱保护的步骤。
[0040]
(7)制备式(v)表示的化合物的己二酸盐的方法,其特征在于,包括采用根据(6)所述的方法制备式(v)表示的化合物,然后使用己二酸盐化的步骤。
[0041]
在本发明中,“钯催化剂”是二价钯催化剂或零价钯催化剂。其实例包括[三(2-甲基苯基)膦]钯(0)。
[0042]
本发明的“钯催化剂”包括在反应体系中通过以下制备的催化剂,例如,通过使单齿膦配体如三苯基膦,三叔丁基膦或三(2-甲基苯基)膦或双齿膦配体如1,1-双(二苯基膦基)甲烷或1,2-双(二苯基膦基)乙烷,作用于钯化合物如氯化钯或乙酸钯。
[0043]
在本发明中,反应可以使用非常少量的钯催化剂进行。基于式(i)化合物的量,所用钯催化剂的量优选为0.5至10mol%,更优选为1至5mol%。所用钯催化剂的量进一步优选为2mol%。
[0044]
可用于本发明的氮原子的保护基(pg)没有特别限制,只要它是降低氮原子对亲电加成反应的反应性的基团即可。例如,可以使用protective groups in organic synthesis(t.w.green和p.g.m.wuts,john wiley&sons,inc.,new york,1991)中公开的保护基团。保护基团优选为叔丁氧基羰基或苄氧基羰基。
[0045]
可用于本发明的溶剂没有特别限制,只要它不抑制涉及由钯催化的c-h活化反应的芳族取代反应即可。其实例包括甲苯,环戊基甲基醚,1,4-二氧六环和二乙二醇二甲醚。溶剂优选为可与水混溶的溶剂,并且其实例包括二乙二醇二甲醚。
[0046]
通过使化合物与酸反应,可以将本发明的式(iii)表示的化合物和式(v)表示的化合物分别转化为盐。
[0047]
盐的实例包括:无机酸盐,包括氢卤化物,如氢氟酸盐,盐酸盐,氢溴酸盐或氢碘酸盐,硝酸盐,高氯酸盐,硫酸盐和磷酸盐;有机酸盐,包括c
1-c6烷基磺酸盐,例如甲磺酸盐、三氟甲磺酸盐或乙磺酸盐,芳基磺酸盐,例如苯磺酸盐或对甲苯磺酸盐,乙酸盐,苹果酸盐,富马酸盐,琥珀酸盐,柠檬酸盐,抗坏血酸盐,酒石酸盐,草酸盐和己二酸盐;和氨基酸盐,例如甘氨酸盐、赖氨酸盐、精氨酸盐、鸟氨酸盐、谷氨酸盐和天冬氨酸盐。
[0048]
当根据本发明的式(iii)表示的化合物或其盐和式(v)表示的化合物或其盐放置在大气中或重结晶时,这些化合物在某些情况下并入水分子并变成水合物。这些水合物也包括在本发明中。
[0049]
当根据本发明的式(iii)表示的化合物或其盐和式(v)表示的化合物或其盐放置在溶剂中或重结晶时,这些化合物在某些情况下吸收某种类型的溶剂并成为溶剂化物。这些溶剂化物也包括在本发明中。
[0050]
发明的有益效果
[0051]
本发明提供了制备3,6-二取代咪唑并[1,2-b]哒嗪衍生物的方法,所述方法使用利用钯催化的c-h活化的芳族取代反应,使用6-氟咪唑并[1,2-b]哒嗪作为起始材料,其中
该方法是一种行业上有用的新方法,其中使用的钯催化剂的量很小,并且具有复杂结构的电子供体取代基的芳基可以高产率被引入。
具体实施方式
[0052]
下面将描述本发明。不应将本发明的反应条件解释为限于以下描述。在本发明中,存在化合物的官能团被合适的保护基团保护的情况。这种官能团的实例包括羟基,羧基和氨基。关于保护基团的类型和引入和除去这些保护基团的条件,可以是参考protective groups in organic synthesis(t.w.green和p.g.m.wuts,john wiley&sons,inc.,new york,1991)中描述的那些。
附图说明
[0053]
[图1]图1显示实施例7中得到的化合物(6)的晶体的粉末x-射线衍射图。纵轴表示衍射强度作为相对线强度(计数),横轴表示衍射角2θ。
[0054]
实施例
[0055]
在下文中,将参考以下实施例更详细地描述本发明。然而,这些实施例不是为了限制本发明的范围。
[0056]
应注意,实施例中使用的缩写具有以下含义。
[0057]
g:克,ml:毫升,l:升,mhz:兆赫兹。
[0058]
在以下实施例中,关于核磁共振(下文称为1h nmr:400mhz)光谱,使用四甲基硅烷作为标准物质,将化学位移值作为δ值(ppm)提供。对于分裂模式,使用以下符号:s,单峰;d,双峰;dd,双双峰;m,多重峰;和br,宽峰。
[0059]
另外,粉末x射线衍射分析装置和分析条件如下。
[0060]
仪器:配有gadds cst的d8 discover,由bruker axs制造
[0061]
x射线源:cukαλ=1.54埃
[0062]
方法:反射法
[0063]
管电压:40kv
[0064]
管电流:40ma
[0065]
扫描范围:2
°
至42
°
[0066]
扫描速度:10
°
/min
[0067]
[实施例1]
[0068]
[(2r)-1-(4-溴苯氧基)丙-2-基]氨基甲酸叔丁基酯(1)
[0069]
[式6]
[0070][0071]
在氮气氛下,加入1-溴-4-氟苯(100g,0.57mol,1当量),n-甲基吡咯烷酮(500ml)和d-氨基丙醇(51.5g,0.69mol,1.2当量),然后在40℃或更低的温度下向其中加入叔丁醇钾(96.1g,0.86mol,1.5当量)。将所得混合物在约65℃的内部温度搅拌3小时并冷却至20℃
或更低。之后,向其中加入乙酸异丙酯(500ml)和水(1000ml),并搅拌所得混合物。静置和分离后,将水层用乙酸异丙酯(500ml)萃取两次,并合并所有有机层。将合并的有机层用水(500ml)洗涤两次,并且将获得的有机层在减压下浓缩至300ml。进一步向其中加入乙醇(1000ml)并将所得混合物在减压下浓缩至300ml的操作重复两次。向该溶液中加入四氢呋喃(200ml),将所得混合物冷却至5℃或更低。将二碳酸叔丁酯(tert-butyl dicarbonate)(162g,0.74mol,1.3当量)溶解在四氢呋喃(100ml)中,并在约2小时内在6℃或更低的温度下将所得溶液滴加到混合物中。将所得混合物在5℃或更低的温度下搅拌1小时,然后升至约20℃并搅拌过夜。向其中加入乙醇(230ml),然后在1.5小时内滴加水(800ml)。将所得混合物在约50℃下搅拌1小时或更长时间,然后逐渐冷却至25℃,并搅拌过夜。过滤沉淀的固体并用乙醇(230ml)和水(270ml)的混合溶液洗涤。将固体在40℃的外部温度下真空干燥,得到标题化合物(1)(170g)。
[0072]
[实施例2]
[0073]
6-氟咪唑并[1,2-b]哒嗪甲磺酸盐(2)
[0074]
[式7]
[0075][0076]
在氮气氛下,苄基三乙基氯化铵(445g,1.95mol,1当量)和6-氯咪唑并[1,2-b]哒嗪(300g,1.95mol,1当量)(可从combi-block等获得)依次加入二甲基亚砜(1500ml)中。进一步向其中加入氟化铯(534g,3.51mol,1.8当量),然后将所得混合物在79℃至81℃的内部温度下搅拌4小时。将混合物冷却至室温,向混合物中加入甲苯(1500ml)和碳酸氢钠(48g,0.59mol,0.3当量),然后向其中加入水(1500ml)。向混合物中加入乙腈(600ml),搅拌所得混合物,然后分离有机层和水层。此外,将用甲苯(1500ml)和乙腈(300ml)的混合溶液萃取该水层的操作重复三次,并合并所有有机层。减压浓缩合并的有机层,将液体体积调节至2400ml。向其中加入用甲苯(150ml)润湿的活性炭(30g)。将所得混合物在约25℃下搅拌1小时,然后过滤并用甲苯(750ml)洗涤。向其中加入乙腈(900ml),然后在22℃至37℃的内部温度下在1小时内滴加甲磺酸(188g,1.95mol,1当量)。将所得混合物在27℃至31℃下搅拌1.5小时,然后过滤沉淀的固体并用甲苯(900ml)洗涤。将固体在减压下在40℃的外部温度下干燥5小时,得到标题化合物(2)(396.9g)。
[0077]
[实施例3]
[0078]
{(2r)-1-[4-(6-氟咪唑并[1,2-b]哒嗪-3-基)苯氧基]丙-2-基}氨基甲酸叔丁酯(3)
[0079]
[式8]
[0080][0081]
在氮气氛下,甲基叔丁基醚(12l),水(2.6l),碳酸钾(691g,5.0mol,1.1当量)和式(2)化合物(1.17kg,5.0mol,1.1当量)相继添加。将所得混合物在19℃的内部温度下搅拌5分钟并静置,然后排出水层。减压浓缩所得有机层,将液体体积调节至7.5l。向其中加入二乙二醇二甲醚(7.5l),再次减压浓缩所得混合物,将液体体积调节至8.25l。向该溶液中,依次加入式(1)化合物(1.5kg,4.54mol,1当量),三(2-甲基苯基)膦(27.7g,0.09mol,0.02当量),碳酸钾(1.26kg,9.12mol)和乙酸钯(20.4g,0.09mol,0.02当量),然后用二乙二醇二甲醚(0.3l)洗涤。将所得混合物在95℃至108℃的内部温度下搅拌9小时,然后在58℃至61℃的内部温度下搅拌11小时。向其中加入纯水(7.5l),将所得混合物温热至内部温度71℃,然后排出水层。向有机层中,加入1-甲基咪唑(1.5l),冷却得到的混合物。将混合物在25℃至30℃下搅拌40分钟,然后在25℃至29℃的内部温度下在1.5小时内间歇地向其中加入水(9l)。将所得混合物在约25℃下搅拌19小时,然后过滤晶体并用二乙二醇二甲醚(3l)和水(3l)的混合溶液洗涤,然后用水(3l)洗涤。将得到的固体在外部温度40℃下减压干燥,得到标题化合物(3)(1.65kg,94.1%(毛重))。
[0082]1hnmr(500mhz,cdcl3):δ=1.32(d,j=7.0hz,3h),1.47(s,9h),4.00(d,j=4.0hz,2h),4.10(brs,1h),4.80(brs,1h),6.87(d,j=7.6hz,1h),7.02-7.08(m,2h),7.92-7.97(m,2h),8.00(s,1h),8.06(dd,j=7.6,6.0hz,1h)
[0083]
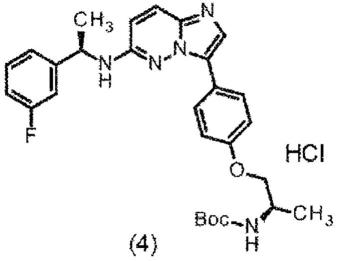
[实施例4]
[0084]
{(2r)-1-[4-(6-{[(1r)-1-(3-氟苯基)乙基]氨基}咪唑并[1,2-b]哒嗪-3-基)苯氧基]丙烷-2-基}氨基甲酸叔丁酯盐酸盐(4)
[0085]
[式9]
[0086][0087]
在氮气氛下,(1r)-1-(3-氟苯基)乙胺(400g,2.87mol,1当量),磷酸三钠(471g,2.87mol,1当量)和式(3)的化合物(1.22kg(净重:1.12kg),3.16mol,1.1当量)依次加入二甲基亚砜(2.4l)中。将该混合溶液加热,并在95℃至99℃的内部温度下搅拌55小时。将溶液冷却,并在24℃的内部温度下向其中加入环戊基甲基醚(4l)和水(8l)。将所得混合物温热
至50℃,并排出水层。之后,将水(4l)加入到剩余的有机层中,再次排出水层。减压浓缩所得有机层,将液体体积调节至4l。使用环戊基甲基醚(0.4l)过滤液体。
[0088]
从所得溶液取出5/8的量,用于随后的反应。向该溶液中依次加入环戊基甲基醚(0.25l),四氢呋喃(3l)和水(0.05l),在23℃内温下向其中加入浓盐酸(74.9g,1.15mol,0.4当量)。将所得混合物在25℃下搅拌1.5小时,然后向其中加入环戊基甲基醚(1.5l)和四氢呋喃(1.5l)的混合溶液。将所得混合物再搅拌1.5小时,然后每小时分3回向其中加入浓盐酸(112g,1.72mol,0.6当量)。将所得混合物在25℃的内部温度下搅拌18小时。过滤沉淀的固体,并用环戊基甲基醚(1.25l),四氢呋喃(1.25l)和水(0.025l)的混合溶液洗涤。将固体在减压下在40℃的外部温度下干燥,得到标题化合物(4)(808.0g)。
[0089]
[实施例5]
[0090]
3-{4-[(2r)-2-氨基丙氧基]苯基}-n-[(1r)-1-(3-氟苯基)乙基咪唑并[1,2-b]哒嗪-6-胺二盐酸盐(5)
[0091]
[式10]
[0092][0093]
在氮气氛下,将式(4)化合物(120.0g)溶解在乙醇(1080ml)中,然后向其中加入用乙醇(60ml)润湿的活性炭(12g)。将所得混合物搅拌1小时,然后过滤并用乙醇(120ml)洗涤。向所得溶液中加入浓盐酸(43.3g),将所得混合物加热,并在65℃至70℃下搅拌4小时。在2小时内将混合物冷却至内部温度20℃,并在该温度下搅拌1小时,然后在1小时内进一步冷却至1℃。将混合物在-1℃至1℃的内部温度下搅拌19.5小时。之后,过滤沉淀的固体并用冷乙醇(240ml)和水(6ml)的混合溶液洗涤。将固体在外部温度40℃下减压干燥,得到标题化合物(5)(100.5g)。
[0094]
[实施例6]
[0095]
3-{4-[(2r)-2-氨基丙氧基]苯基}-n-[(1r)-1-(3-氟苯基)乙基咪唑并[1,2-b]哒嗪-6-胺(v)
[0096]
在氮气氛下,将式(5)化合物(75.5g,0.17mol),乙醇(604ml)和水(604ml)混合在一起,然后升温至内部温度50℃以溶解化合物。在内温50℃下用3分钟向其中加入25%氢氧化钠水溶液(68.1g)。之后,将所得混合物在1.5小时内冷却至1℃的内部温度并搅拌18.5小时。过滤沉淀的固体并用乙醇(151ml)和水(151ml)的冷混合溶液洗涤。将固体在减压下在40℃的外部温度下干燥,得到标题化合物(v)(58.8g)。
[0097]
[实施例7]
[0098]
3-{4-[(2r)-2-氨基丙氧基]苯基}-n-[(1r)-1-(3-氟苯基)乙基咪唑并[1,2-b]哒嗪-6-胺己二酸盐(6)
[0099]
[式11]
[0100][0101]
在氮气氛下,将乙醇(90ml)加入到式(v)化合物(30.0g,1当量)中,并将温度升至50℃以溶解化合物,然后过滤。将己二酸(11.4g,1.1当量)溶解在乙醇(75ml)和水(75ml)的混合溶液中,然后过滤。将滤液加入至3-{4-[(2r)-2-氨基丙氧基]苯基}-n-[(1r)-1-(3-氟苯基)乙基咪唑并[1,2-b]哒嗪-6-胺溶液。向其中加入水(54ml),在内温27℃下加入30.0mg(0.1重量%)晶种*,之后搅拌18小时。在约41℃的内部温度下在1.3小时内向其中加入水(306ml),然后将所得混合物搅拌2小时。此外,将混合物在1.5小时内冷却至-1℃的内部温度并搅拌16.5小时。过滤晶体并用冷的乙醇(18ml)和水(42ml)的混合溶液洗涤。将晶体在40℃的外部温度下减压干燥,得到标题化合物(6)的晶体(37.2g)。所得晶体的xrd图显示在图1中。
[0102]
*当反应溶液长时间搅拌时,晶体自发沉淀。然而,此处,为了减少晶体沉淀所需的时间,加入先前在同样实验中获得的晶体作为晶种。
[0103]
[参考例]
[0104]
{(2r)-1-[4-(6-氯咪唑并[1,2-b]哒嗪-3-基)苯氧基]丙-2-基}氨基甲酸叔丁酯(7)
[0105]
[式12]
[0106][0107]
在非专利文献1中公开的条件下使6-氯咪唑并[1,2-b]哒嗪和式(1)化合物反应(乙酸钯,0.1当量;三苯基膦,0.2当量;碳酸钾,2当量;甲苯,110℃;24小时)。结果,式(7)化合物在hplc上的反应率约为1.4%。
[0108]
与非专利文献1中公开的事实相反,显示当使用6-氯咪唑并[1,2-b]哒嗪作为起始材料时,具有复杂结构的给电子取代基的芳基的引入导致产率大幅减少。相反,在使用6-氟咪唑并[1,2-b]哒嗪作为起始材料的实施例3中,尽管所用钯催化剂的量是参考例的五分之一,但显示出高反应速率,这表明本发明的优异效果。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
