人pole基因突变的新型测定方法及试剂
技术领域
1.本发明属于诊断学领域,更具体地,本发明涉及人pole基因突变的新型测定方法及试剂。
背景技术:
2.根据目前中国癌症患者人数统计,每天超过1万人被确诊为癌症,每分钟就有7.5人被确诊为癌症,且数据还有持续上升的趋势。恶性肿瘤发病率每年保持在约3.9%的增幅,死亡率每年保持在2.5%的增幅。随着肿瘤细胞分子生物学的发展,癌症免疫治疗己开始进入临床应用,选择合适的患者进行免疫治疗是其成功的关键。
3.dna聚合酶ε是具有校正功能的dna聚合酶,其催化亚基由pole基因编码。此酶中的核酸外切酶区(exonuclease domain of pole,pole-exo),具有3
’→5’
核酸外切酶活性,能够及时识别并切除复制过程中产生的错误碱基。编码pole核酸外切酶区的基因突变将导致校正功能缺失,导致突变的基因在细胞中大量累积。近年来在子宫内膜样癌、结肠癌等多种肿瘤中检测出pole突变,表现出独特的分子表型,预后较好。故检测pole基因核酸外切酶区是否存在突变可以判断肿瘤预后的有用指标。
4.pole体细胞突变常发生在约1~2%的结直肠癌(crc)中和7~12%的子宫内膜癌(ec)中,并且在脑,胰腺,卵巢,乳房,胃的超突变肿瘤以及子宫癌肉瘤中被检测到,最常见的突变类型是p286r、v411l、s297f、a456p、s459f,此外f367s、l424v、l424i、p346r、m444k也较为常见。这些突变直接或间接的影响了dna聚合酶ε核酸外切酶区域的校正功能。
5.pole基因突变的肿瘤细胞由于积累了大量的基因变异,导致产生的蛋白更容易被患者的免疫系统发现,故可增强患者的免疫应答。研究人员在肿瘤免疫组化染色中发现pole突变的肿瘤中存在大量的cd8 阳性的浸润淋巴细胞和细胞毒性t淋巴细胞标志物,pd-1》90%,并在肿瘤微环境中鉴定出pd-l1的大量表达,表明具有pole突变的肿瘤可能是靶向pd1-pdl1抗体药物的候选者。己有临床研究报道携带pole基因突变的癌症患者接受针对pd-1的免疫治疗具有良好的疗效。
6.此外,鉴于pole基因突变位点的多样性,一些实验室和研究机构中,pole基因的变异研究也是被聚焦研究的对象,从而为临床试剂的开发优化奠定基础。
7.目前针对pole基因突变检测方法包括高通量测序ngs、数字pcr(dpcr)。ngs方法检测步骤繁杂、试剂费用昂贵、结果判读依赖于生物信息学分析计算,检测周期长:数字pcr方法检测灵敏度可达0.1%,但其检测步骤较多、试剂费用较实时荧光pcr昂贵很多、结果判读也依赖于专用软件、对阴性/阳性区间的划分主要依赖于针对图形的主观判断。ngs和数字pcr仪器试剂昂贵,同时对操作人员也有很高要求。
技术实现要素:
8.本发明的目的在于提供一种人pole基因突变的新型测定方法及试剂。
9.在本发明的第一方面,提供一种检测pole基因多位点突变的试剂盒,所述突变包
括pole基因第857、890、1100、1231、1270、1307、1331、1366、1376位突变;该试剂盒中包括:(1)扩增pole基因的所述突变位点区域的特异性前引物和后引物,所述的特异性前引物和后引物扩增包含基因突变位点在内的扩增产物的长度为50~200bp(较佳地为50~150bp;例如73bp,81bp,102bp,119bp);所述特异性前引物或后引物与突变型基因位点及邻近序列互补,较佳地前引物或后引物3’末端碱基与相应突变位点碱基互补;(2)特异性针对扩增产物的探针,所述的探针携带可检测标记;(3)block引物,其靶向结合pole基因突变位点对应的野生型位点,所述pole基因突变位点为857、890、1100、1231、1366、1376。
10.在一个优选例中,所述pole基因多位点突变包括:c.857c》g、c.890c》t、c.1100t》c、c.1231g》t、c.1231g》c、c.1270c》g、c.1270c》a、c.1307c》g、c.1331t》a、c.1366g》c、c.1376c》t。
11.在另一优选例中,所述block引物包括:seq id no:3,seq id no:6,seq id no:9,seq id no:14,seq id no:28,seq id no:31所示核苷酸序列的block引物。
12.在另一优选例中,所述的可检测标记是荧光标记。
13.在另一优选例中,所述的特异性前引物的长度为10-25bp;较佳地为15-22bp(如16、17、18、19、20、21bp)。
14.在另一优选例中,所述的特异性后引物的长度为10-25bp;较佳地为15-22bp(如16、17、18、19、20、21bp)。
15.在另一优选例中,对于第857位突变,前引物和后引物分别为seq id no:1和seq id no:2所示核苷酸序列的引物;对于第890位突变,前引物和后引物分别为seq id no:5和seq id no:2所示核苷酸序列的引物;对于第1100位突变,前引物和后引物分别为seq id no:7和seq id no:8所示核苷酸序列的引物;对于第1231位突变,前引物为seq id no:11和seq id no:12所示核苷酸序列的引物,后引物为seq id no:13所示核苷酸序列的引物;对于第1270位突变,前引物为seq id no:16和seq id no:17所示核苷酸序列的引物,后引物为seq id no:18所示核苷酸序列的引物;对于第1307位突变,前引物和后引物分别为seq id no:20和seq id no:21所示核苷酸序列的引物;对于第1331位突变,前引物和后引物分别为seq id no:23和seq id no:24所示核苷酸序列的引物;对于第1366位突变,前引物和后引物分别为seq id no:26和seq id no:27所示核苷酸序列的引物;和/或,对于第1376位突变,前引物和后引物分别为seq id no:30和seq id no:27所示核苷酸序列的引物。
16.在另一优选例中,对于第857位突变,探针的核苷酸序列如seq id no:4所示;对于第890位突变,探针的核苷酸序列如seq id no:4所示;对于第1100位突变,探针的核苷酸序列如seq id no:10所示;对于第1231位突变,探针的核苷酸序列如seq id no:15所示;对于第1270位突变,探针的核苷酸序列如seq id no:19所示;对于第1307位突变,探针的核苷酸序列如seq id no:22所示;对于第1331位突变,探针的核苷酸序列如seq id no:25所示;对于第1366位突变,探针的核苷酸序列如seq id no:29所示;和/或,对于第1376位突变,探针的核苷酸序列如seq id no:29所示。
17.在另一优选例中,所述试剂盒中还包括选自下组:核酸提取试剂;dna扩增试剂;pcr扩增缓冲液;核酸序列分析软件;和/或,说明操作方法的使用说明书。
18.在本发明的另一方面,提供所述的试剂盒的用途,用于检测pole基因多位点突变;所述突变包括pole基因第857、890、1100、1231、1270、1307、1331、1366、1376位突变。
19.在一个优选例中,所述的用途为非诊断性的用途。例如,应用于在实验室中分析pole的变异情况,或应用于不以诊断为目的的辅助性样本参数测定。
20.在本发明的另一方面,提供一种检测pole基因多位点突变的方法,所述突变包括pole基因第857、890、1100、1231、1270、1307、1331、1366、1376位突变;所述方法包括:(i)以含有待测基因的样品为模板,以前面任一所述的试剂盒中的试剂进行pcr扩增;(ii)分析pcr扩增产物,确定待测样品中pole基因第857、890、1100、1231、1270、1307、1331、1366或1376位的突变类型。
21.在一个优选例中,所述的方法为非诊断性的方法。例如,应用于在实验室中分析pole的变异情况,或应用于不以诊断为目的的辅助性样本参数测定。
22.在另一优选例中,根据靶位点和内参基因的ct值的差(δct值)来判断pole基因的突变型,δct值越小(如小于8、7、6、5、4、3、2或1),表明该位点突变型的含量越大。
23.在另一优选例中,所述的内参基因是β-珠蛋白基因(β-globin)。
24.在另一优选例中,所述的β-globin基因的扩增引物包括seq id no:32和seq id no:33所示序列的引物。
25.在另一优选例中,所述的β-globin基因的探针为seq id no:34所示序列的探针。
26.在另一优选例中,所述的β-globin基因的引物和探针可掺入所有靶位点的扩增体系一起扩增,作为内控验证样本质量;也可单独做一个孔的扩增做为外控,参与计算δct。
27.本发明的其它方面由于本文的公开内容,对本领域的技术人员而言是显而易见的。
附图说明
28.图1、pole突变检测的临床样本中1号样本的sanger测序结果。
29.图2、pole突变检测的临床样本中2号样本的sanger测序结果。
30.图3、pole突变检测的临床样本中3号样本的sanger测序结果。
31.图4、pole突变检测的临床样本中4号样本的sanger测序结果。
32.图5、pole突变检测的临床样本中5号样本的sanger测序结果。
具体实施方式
33.鉴于pole基因突变位点的多样性以及本领域中对其突变鉴定尚需稳定可靠的方法和试剂的情况,本发明人在深入的研究和试验工作中,不断对其突变位点的检测技术进行优化和改良。在这些工作的基础上,本发明提出了一组适用于进行pole基因多位点突变检测的方法和试剂,本发明的技术方案,可以一次性同时检测pole基因多达11种突变型,亦可实现对每一种突变类型进行区分,不仅检测特异性和灵敏度俱佳,且操作便捷、耗时短、成本低。
34.如本文所用,所述的“pole基因多位点突变”是指对应于pole蛋白的第286位、297、367、411、424、436、444、456、459位的氨基酸残基,编码其的基因的密码子发生突变,较佳地,在pole基因的第857、890、1100、1231、1270、1307、1331、1366、1376位发生碱基的突变。更为具体地,,所述突变包括:第857c》g、890c》t、1100t》c、1231g》t、1231g》c、1270c》g、1270c》a、1307c》g、1331t》a、1366g》c、1376c》t。
35.如本文所用,所述的“前引物”与“正向引物”可互换使用。通常,利用该“前引物”与“后引物”进行pcr扩增,获得一个具有合理长度的扩增产物,例如长度为50-200bp;较佳地为50-120bp。
36.如本文所用,所述的“后引物”与“反向引物”可互换使用。
37.如本文所用,“互补”是指核苷酸的序列之间(如本发明中前引物序列与pole基因突变位点及邻近位点的序列之间)可以以一种可预见的方式发生相互作用,如形成二级结构(如茎环结构)。通常,两条“基本上互补”的核苷酸序列互相之间至少有70%的核苷酸是互补的;更优选的,至少有80%或90%的核苷酸是互补的。本发明中,两条足够互补的分子之间可以具有1-2个(较佳地为1个)不配对的核苷酸;例如将不配对的核苷酸位置设置于3’端的倒数第2或3位。
38.如本文所用,碱基的“匹配”、“配对”或“完全互补”是指两条核苷酸序列中相应的碱基构成了键(如氢键)的连接,例如“a”与“t”之间可形成键。两条序列中足够多核苷酸的“配对”可使得两条序列发生互补。
39.如本文所用,碱基的“基本上匹配”是指两条核苷酸序列中绝大多数的碱基构成了键(如氢键)的连接,存在个别(例如1个)碱基的“错配”。
40.如本文所用,所述的碱基“错配”是指两条核苷酸序列中,存在位置关系相应的两个碱基,它们之间不构成键(如氢键)的连接。
41.如本文所用,“待测样本”或“待测核酸(dna)样本”是指待检测的核酸样本,其中含有一种核酸或多种核酸,需要了解其中是否存在目标核酸的目标突变。
42.本发明中,以pole基因多突变位点共同考虑为检测对象,将突变检测位点设计于引物(m)(包括前引物或后引物,根据检测的实际情况而设计)上,综合考虑突变位点的特点以及整体反应体系,提出可以一次性同时检测pole基因多达11种突变型的一些优选的突变检测方案。
43.基于本发明人的新发现,本发明提供了一种检测pole基因多突变的试剂盒,所述突变包括pole基因第857、890、1100、1231、1270、1307、1331、1366、1376位突变;所述试剂盒包括:(1)特异性扩增pole基因多突变位点区域的前引物和后引物,所述的前引物和后引物扩增包含基因突变位点在内的扩增产物的长度为50-150bp;所述前引物与突变型基因位点及邻近序列互补(优选全部互补),较佳地其3’末端碱基与pole基因中相应的突变位点的碱基互补;(2)特异性针对扩增产物的探针(p),所述的探针携带可检测标记;以及(3)block引物(b),其靶向结合pole基因突变位点对应的野生型位点,所述pole基因突变位点为857、890、1100、1231、1366、1376。
44.一些复杂的待测样本,例如组织和血液样本中,除了突变型dna,还含有大量的野生型dna,对于检测的特异性造成了干扰。本发明中,针对pole基因多突变位点中的部分位点,应用竞争性block引物来排除野生型dna的干扰,对于提高检测的特异性是特别有效的。block引物与野生型基因完全互补,而与突变基因部分互补,在有野生型基因存在的情况下,block寡聚核苷酸能够封闭野生型基因,防止野生型基因扩增造成的假阳性。
45.本发明人在深入研究和比较实验中发现,针对部分位点的block引物的应用,有利于提高检测的灵敏度,所述的部分位点为857、890、1100、1231、1366、1376。同时,本发明人也发现,其它一些位点上,block引物并非是必需的,鉴于block引物实质上会增加反应系统
的复杂性,有些block引物的应用会导致反应的不确定性,或减少突变型的扩增,反而起到反作用。基于本发明人的这些发现,本发明中,所述的block引物优选地为配合于pole基因突变位点为857、890、1100、1231、1366、1376的block引物。
46.作为本发明的优选方式,可以对所述block引物(b)进行修饰;较佳地,在其3’端进行修饰,从而阻滞block引物的延伸。多种可阻滞核苷酸序列延伸的修饰方式可被应用于本发明中,例如,所述修饰可包括但不限于c3spacer、磷酸、氨基。
47.本发明中,所述的前引物或后引物的长度可以是10-25bp;较佳地为15-22bp。本发明人考察不同长度的后引物对于突变基因与野生基因的分辨能力,长度为15-22bp的引物长度是相对理想的。本发明人在研究中也观察到,当进行复杂体系的检测时,优化引物以获得适当的扩增产物,对于实现良好的特异性和灵敏度是重要的。经过反复的实验和比较、结合多突变位点的特点,本发明提供了一系列优化的引物组,具体地其被记载于本发明的表2中。
48.作为本发明的优选方式,所述突变引物的3’端第1个脱氧核苷酸可以是锁核苷酸,从而增强对突变型模板的结合能力。
49.本发明中,应用了特异性针对扩增产物的探针来对扩增产物进行特异性检测。较佳地,所述的探针是带有可检测标记的探针,如荧光探针。
50.作为一种优选方式,所述的探针连接有适合于甄别突变型别的荧光基团。探针的前端标记有荧光基团,另一端标记有淬灭基团,其中淬灭基团可淬灭荧光基团发出的荧光。当pcr扩增反应进行时,利用聚合酶的正向外切活性将带有荧光基团的碱基切离,游离后的荧光基团不再受淬灭基团的影响,在激发光的作用下可发射一定波长的荧光信号。随着pcr产物的不断积累,荧光信号不断地增强,从而可以检测到目标扩增产物的存在。
51.所述探针的5’端可进行荧光基团的修饰,所述修饰包括但不限于fam、hex、tet、vic、joe、rox、tamra、cy3、cy5等,从而可产生荧光。
52.与探针的5’端的修饰相应地,所述探针的3’端可进行修饰,所述修饰包括但不限于tamra、bhq1、bhq2、bhq3等,用于淬灭荧光。
53.作为一种可选择的方式,所述突变引物的3’端倒数第2和第3个脱氧核苷酸的碱基可以变动,包括但不限于腺嘌呤a、胸腺嘧啶t、胞嘧啶c、鸟嘌呤g、尿嘧啶u和次黄嘌呤i等。用于减少对野生型模板的结合能力。
54.经过反复的实验和比较、结合多突变位点的特点,本发明人在提供了一系列优化的引物组的同时,也优化设计了相应的探针,具体地其被记载于本发明的表2中。
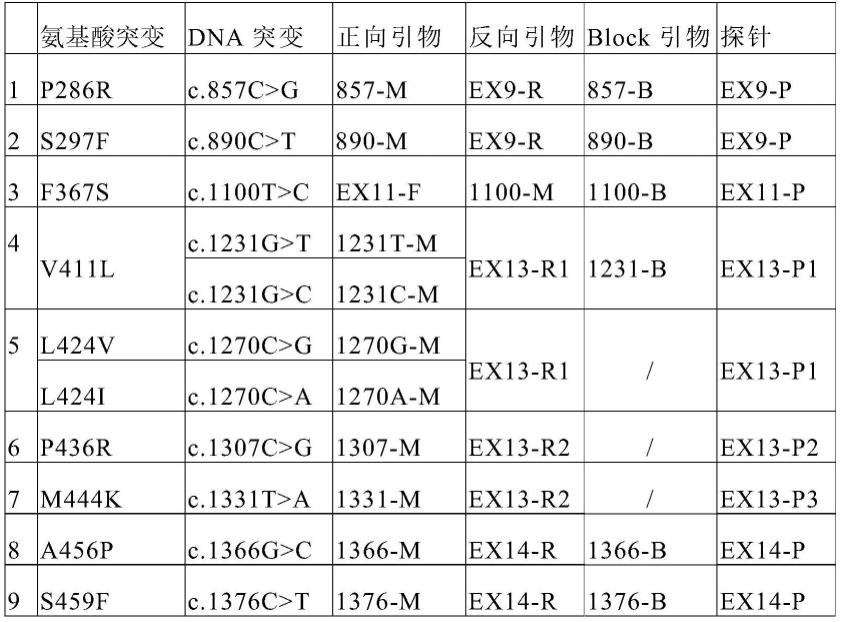
55.本发明的具体实施例中,将突变位点置于突变引物的3’端,可以实现引物对突变型模板的延伸和对野生型模板的阻滞。dna聚合酶延伸引物后遇到探针时可将其水解,从而报告荧光。而block引物和野生型模板互补,其3’端进行了修饰以阻断延伸,可与突变引物竞争野生型模板,进一步提高特异性。本发明的实施例中,相应于所述pole基因的突变检测位点的引物及探针组合如表1,其更具体的信息则被记载于表2中。
56.表1
[0057][0058]
本发明所述的各组引物、探针及block引物组合可以单独一管检测,也可以对两组或多组混合在一管内检测。混合在一管内检测时,可以对所有探针设计同一种荧光基团,此时对突变位点不做区分;亦可对不同组的探针设计不同的荧光基团,此时可通过仪器不同荧光通道的读数来区分不同的突变位点。
[0059]
本发明所述试剂盒中,还可包括(但不限于):dna扩增试剂,pcr扩增缓冲液,金属离子如镁离子等。以及还可包括说明操作方法的使用说明书,从而有利于本领域技术人员方便地实施检测。
[0060]
作为本发明的优选方式,根据所采用的实时荧光pcr方法,本发明的试剂盒中还可以包括内参基因。
[0061]
本发明还提供了一种检测pole基因多突变的方法,所述方法包括:(i)以待测基因为模板,以所述的试剂盒中的试剂进行pcr扩增;(ii)分析pcr扩增产物,确定待测样品中pole基因突变类型;较佳地,确定待测样品中pole基因第857、890、1100、1231、1270、1307、1331、1366或1376位的突变类型。
[0062]
作为本发明的一种检测方式,本发明人按突变型和野生型差距最大化来优化体系,提供一种用于检测pole基因突变的方法,其包括以下步骤:
[0063]
(1)针对pole基因突变位点,设计特异性引物、探针及block引物。
[0064]
(2)提取检测样本中dna,检测样本包括新鲜病理组织、石蜡包埋组织等。
[0065]
(3)建立实时荧光pcr扩增反应体系:
[0066]
(4)根荧光仪显示的ct值,计算δct值,根据cutoff判断检测结果:当δct《cutoff时,判为阳性,否则判为阴性或低于检测限。
[0067]
作为本发明的优选检测方式,将本发明的技术方案应用于临床实践中,使用内参基因作为标准,野生型靶位点和内参基因具有较大的δct,突变型则较小。较佳地所述的内
参基因是β-珠蛋白基因(β-globin)。更佳地所述的β-globin基因的扩增引物包括seq id no:32和seq id no:33所示序列的引物,所述的β-globin基因的探针为seq id no:34所示序列的探针。探针荧光基团选用hex/vic/tet等,与fam或cy5等靶位点探针的荧光做区分。所述的β-globin基因的引物和探针可掺入所有靶位点的扩增体系一起扩增,作为内控验证样本质量;也可单独做一个孔的扩增做为外控,参与计算δct。
[0068]
本发明的技术方案,可以适用于绝大多数的复杂样本,包括血液标本,ffpe样本等。
[0069]
本发明的方法可以被应用于非诊断性的方法。例如,仅在实验室中应用该方法分析pole基因的变异情况,进行研究分析,这种实施方式不面对患者,不给出诊断结果,属于不以诊断为目的的方法。鉴于pole具有多种突变的特性,本发明的试剂盒及其测试方法,显然是研究机构/实验室所需要应用的。
[0070]
与现有技术相比,本发明的有益效果在于:
[0071]
(1)本发明优化了pole基因突变位点的检测试剂,可以一次性同时检测pole基因11种突变型,亦可对每一种突变类型进行区分。
[0072]
(2)本发明的方法仅需涉及实时荧光pcr反应体系的配制、加样和上机,操作步骤非常简单,检测速度快、周期短,提取dna后可在90分钟内获得结果:判读过程简单,无需其他专业数据分析处理软件,也无需通过图形进行阴性/阳性区间的人为主观判断,非常易于判读结果;检测试剂费用相对于ngs、数字pcr等需要使用昂贵的检测仪器配套专用试剂而言要低很多。
[0073]
(3)本发明的检测方法和试剂的灵敏度高,可达0.1%;检测特异性强,可耐受3.3μg正常人基因组dna。在突变点多达11种的情况下,各个引物/探针能够良好地兼容,实现高特异性和灵敏度,这是出乎意料的。
[0074]
(4)本发明的优选方案中,简化了所用的试剂的数量,将针对多个位点的检测试剂进行简并设计。包括在针对s297f突变的试剂中,将探针和反向引物与针对p286r的探针和引物共用;在针对m444k突变的试剂中,将反向引物与针对p436r的反向引物共用,从而有效地降低了试剂成本,最大化降低反应体系的复杂性。
[0075]
(5)采用实时荧光pcr技术,pcr反应结束后无需开盖,有效避免了核酸气溶胶污染。
[0076]
(6)相对于ngs、数字pcr方法,本发明中的实时荧光pcr方法和试剂盒检测周期更短、检测试剂成本更低。
[0077]
(7)pole基因作为一种突变位点复杂多变的基因,本发明可实现一次性检测pole基因的11种dna突变(对应9种氨基酸突变),所述突变位点囊括了常见的和较少见的位点。
[0078]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如j.萨姆布鲁克等编著,分子克隆实验指南,第三版,科学出版社,2002中所述的条件,或按照制造厂商所建议的条件。
[0079]
荧光定量pcr方法
[0080]
定量pcr检测方法如下:
[0081]
缓冲液融化,振荡均匀;dntp融化后需放入冰块中;taq酶备用;模板(待测者的基
因组dna)完全融化后,放入冰块中。
[0082]
pcr反应体系如下(总20μl):
[0083][0084]
pcr操作过程如下:
[0085]
(1)制备预混液:先加入ddh2o,然后加入引物、探针、block引物,缓冲液,加入dntp,最后加入taq dna聚合酶,立即振荡混和均匀;
[0086]
(2)把模板加入到pcr薄壁管的底部,最后一管加入ddh2o作为阴性对照;
[0087]
(3)将pcr预混液分装到每一个pcr反应管中;
[0088]
(4)全部溶液加完后,离心以保证管壁上没有残留pcr反应液。
[0089]
pcr反应的一般程序:
[0090]
95℃5min,变性;
[0091]
95℃10sec,53℃15sec,72℃35sec,进行45个循环。
[0092]
22℃保存。
[0093]
除非另外说明,后续的实施例采用该荧光定量pcr方法。
[0094]
实施例1、引物的选择
[0095]
本发明人在实验中发现,由于需要一次性同时检测pole基因多种突变型,检测体系非常复杂,导致检测的准确性难以确保,背景值大,假阴性、假阳性问题较难克服。
[0096]
为了找到检测效果相对理想的检测试剂,本发明人基于pole基因及其突变位点的特点进行了深入的研究和实验工作,考虑了多位点/多方式的引物及探针设计策略,一一考察检测的准确性(包括背景干扰、假阳性假阴性等等因素)。
[0097]
经过一些比较,本发明人将突变位点置于突变引物的3’端。本发明人发现,如此设计结合后,引物对突变型模板的延伸和对野生型模板的阻滞作用是理想的。同时,本发明人发现,在一些特定区域进一步设计靶向结合的block引物,可以进一步提高检测效率。
[0098]
本实施例优化设计后的引物和探针序列见表2。包括下列9组引物(
“‑
m”:突变型引物,
“‑
r”:反向引物)、探针(
“‑
p”)及block(
“‑
b”),用于同时检测pole基因中9个dna位点11种类型的突变。除了第3组引物组中突变位点相应碱基位于后引物,其它组引物中位点相应碱基位于前引物。
[0099]
表2
[0100]
[0101][0102]
[0103]
实施例2、block引物的影响
[0104]
如前所述,本发明人在检测试剂的设计中,考虑了block引物的应用。然而,本发明人在反复实验和分析中发现,针对所有位点的检测试剂来运用block引物并非是最恰当的。鉴于block引物实质上会增加反应系统的复杂性,有些block引物的应用会增加反应的不确定性,或减少突变型的扩增,反而起到反作用。因此本发明人针对每一位点深入分析,在针对部分位点的检测试剂中,应用block引物。
[0105]
反应模板的制备:将突变型质粒稀释至100拷贝/μl,将野生型质粒稀释至105拷贝/μl,分别按照前面所述的“荧光定量pcr方法”进行测定。
[0106]
对于部分位点,相同拷贝数下,使用与不使用block引物或其浓度高低不同时,pcr检测效果,浓度高低的数据如表3。
[0107]
表3
[0108]
[0109][0110]
根据上表可见,对于本发明人认为适合以block引物配合的位点而言,随着block浓度增加野生型的ct增加明显,突变型ct值略有增加,两者差距扩大(扩大ct值差距的效果每个突变位点不一)。
[0111]
而对于c.1270c》a、c.1270c》g、c.1307c》g、c.1331t》a的位点,尽管本发明人在实验中也反复验证了多种可能的block试剂,但是综合考虑整体反应体系、成本及效率而言,实际上它们并非必要。
[0112]
实施例3、灵敏度的考察
[0113]
本实施例中,本发明人分别将不同pole突变型质粒混合于纯野生型的质粒中,以考察检测的灵敏度。
[0114]
反应模板的制备:将100拷贝的不同突变型质粒分别和105拷贝的野生型质粒混合;按照前面所述的“荧光定量pcr方法”进行测定,并且如实施例2所述针对部分位点应用block引物。
[0115]
结果如表4。
[0116]
表4
[0117][0118]
结果显示,将100拷贝的突变型质粒和105拷贝的野生型质粒混合,作为0.1%突变丰度的样本进行测试,所有突变位点均能检出,能与纯野生型区分。且所有含突变位点的野生型样本ct均大于35,突变样本ct小于35。
[0119]
上结果也可确认,本发明的方法对各个pole突变位点的灵敏度均可达到0.1%(105拷贝野生型中的100拷贝)。
[0120]
实施例4、混合检测实例
[0121]
本实施例中,将表2中所设计的所有探针修饰fam基团,所有引物、探针、block混合在一管内进行检测。
[0122]
将11种不同突变型质粒(各100拷贝)分别和105拷贝的野生型质粒混合,作为0.1%突变丰度的样本进行检测。按照前面所述的“荧光定量pcr方法”进行测定。结果如表5。
[0123]
表5
[0124]
检测样本ct值c.857c》g30.72c.890c》t34.15c.1100t》c34.02c.1231g》c33.69c.1231g》t33.76c.1270c》a33.96c.1270c》g31.73
c.1307c》g33.82c.1331t》a32.02c.1366g》c32.85c.1376c》t32.95纯野生型36.17
[0125]
所有含量在0.1%的11种突变型质粒的ct均小于35,而野生型则大于35;所有突变体均能够与纯野生型质粒区分。
[0126]
实施例5、pole突变检测的临床样本检测
[0127]
本实施例中,利用前述涉及的试剂进行临床样本检测。
[0128]
获取子宫内膜癌患者手术后的肿瘤组织,提取dna后进行实验。以β-globin作为内参基因。
[0129]
选用的内参基因是β-珠蛋白基因β-globin,其引物、探针序列如下:
[0130]
β-globin上游:acccttaggctgctggtgg(seq id no:32);
[0131]
β-globin下游:ggagtggacagatccccaaa(seq id no:33);
[0132]
β-globin探针:ctacccttggacccagaggttctttgagtc(seq id no:34)。
[0133]
检测结果的判定方式:以感兴趣突变位点的ct值减去内参的ct值为δct;δct大于10判定为阴性,小于等于10判定为阳性。
[0134]
结果如表6。
[0135]
表6
[0136][0137][0138]
表中,内参基因指β-globin。ct空白指没有扩增曲线出现。
[0139]
作为对比和确认,对上述样本的部分dna片段进行sanger测序。
[0140]
1号样本的sanger测序结果如图1;根据该结果,可判定存在c.857c》g突变。
[0141]
2号样本的sanger测序结果如图2;根据该结果,可判定存在c.857c》g突变。
[0142]
3号样本的sanger测序结果如图3;根据该结果,可判定存在c.1231g》t突变。
[0143]
4号样本的sanger测序结果如图4;根据该结果,可判定存在c.890c》t突变。
[0144]
5号样本的sanger测序结果如图5;根据该结果,可判定存在c.857c》g突变。
[0145]
根据上述可见,sanger测序结果与表6的测定结果完全相符,准确性达到100%。
[0146]
实施例6、本发明的方法的检测效率及成本分析
[0147]
对于临床实际检测而言,一种检测方法的是否可规模化应用,除了考虑其准确性、
灵敏度,背景干扰等方面外,另外检测的效率(节约时间)以及检测的成本也是非常重要的方面。
[0148]
本发明人在优化获得了如前实施例所述的较优的检测试剂后,比较了本发明的检测试剂及方法与其它一些可用于pole基因突变体检测的方法的检测灵敏度、特异性、准确度,以及耗费时间和所需成本。
[0149]
运用不同的方法(本发明的方法根据上述,其它几种方法根据相应实验仪器、运用的检测引物、探针的设计运用根据本领域已有标准),针对pole分析所有11种突变型,结果如表7。
[0150]
表7
[0151]
检测方法仪器灵敏度特异性准确度耗费时间成本(试剂)sanger测序sanger测序仪15%最高金标准1-2天数十元ngsngs测序仪5%高高5天以上约一千元数字pcr数字pcr仪0.1%中中1天数百元本发明qpcr仪0.1%高高1天数十元
[0152]
根据上述可见,sanger测序是目前所有测序方法的金标准,优点是成本低、结果直观;但是其缺点是灵敏度很低,对丰度小于15%的突变不能测得。
[0153]
ngs测序是目前常用的突变检测手段,优点是灵敏度较高;但是其缺点是成本大、耗费时间长,数据处理复杂,需要依靠专业人员进行分析,常规实验室或医疗场所难以具备此种仪器。
[0154]
数字pcr是新近发展的技术,优点是灵敏度高,操作时间短;但是其缺点是成本较高,数据处理需要依靠专业人员和软件,常规实验室或医疗场所难以具备此种仪器。
[0155]
本发明的检测方法同数字pcr有相似之处,同样基于荧光pcr,优点是时间短、灵敏度高,且试剂成本、仪器成本远小于数字pcr,其结果可通过简单的加减运算完成,适用于常规实验室以及医疗场所的运用。
[0156]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。